Nose to Brain Delivery of Astaxanthin–Loaded Nanostructured Lipid Carriers in Rat Model of Alzheimer’s Disease: Preparation, in vitro and in vivo Evaluation

Introduction
Alzheimer’s disease (AD) is a chronic, neurodegenerative, multifactorial and progressive brain disorder, which is considered the most common cause of dementia worldwide.1,2 AD represents a public health problem, as the commonly prescribed medications are only symptomatic that do not stop the progressive pathology of the disease.3 At the pathological level, AD is characterized by the presence of extracellular amyloid plaques, formed by amyloid beta (Aβ) deposits, and intracellular neurofibrillary tangles (NFTs), formed by tau protein; these lesions induced neural cell death, which is reflected in memory impairment.4 In addition to the involvement of oxidative stress in the brain, which induces mitochondrial dysfunction, inflammatory activation of glial cells and dysfunction in neurotransmitters release such as acetylcholine, which is associated with cognitive deterioration.5
Astaxanthin (AST) is a lipophilic second-generation antioxidant with anti-inflammatory and antiapoptotic properties, and has the potential to target multiple pathways, which play key roles in the development and progression of AD.6 AST (3,3−-dihydroxy-β,β−-carotene-4,4−-dione) has superior antioxidant activity compared to β-carotene and vitamin E.7,8 AST has the ability to protect the tissues from oxidative stress, scavenge reactive oxygen species (ROS), inhibit lipid peroxidation, attenuate microglial activation, the release of pro-inflammatory cytokines, mitochondrial dysfunction, suppress the amyloidogenic pathway, protect neuronal integrity and exert antiaging effects.8,9 Astaxanthin has the capacity to cross the blood brain barrier (BBB) and research evidence suggested the neuroprotective potential of AST in neurodegenerative diseases.6,10 AST is a natural red xanthophyll carotenoid and is mainly derived from algae, yeast, and marine life such as lobster, shrimp, salmon and crab.11 The natural origin of AST used in this study is the green micro-alga Haematococcus pluvialis. All-trans-AST isomer is the predominant form of AST in nature, as it is more stable that cis-AST, 3S,3S−-AST is a more powerful antioxidant than the other stereoisomers, and it mostly occurs in fatty acid ester form.12,13 AST is commercially available as an antioxidant food supplement (4 mg daily) produced from Haematococcus pluvialis. The European Food Safety Authority (EFSA) advises an acceptable daily intake (ADI) of 0.034 mg/kg/day astaxanthin (2.38 mg/day in a 70-kg human).14 Pharmacokinetic data of AST are available in the literature from several previous studies after administration of different doses of AST.15–17 It is reported that AST can accumulate in the brain of rats.18,19 In Iwamoto et al’s20 study, 24 healthy volunteers consumed AST at doses of 1.8, 3.6, 14.4, and 21.6 mg/day for 14 days without any adverse events.20 Like other carotenoids, AST has low oral bioavailability due to its high lipophilicity.11
Intranasal (IN) administration is a convenient, highly vascular, and non-invasive alternative route for drug delivery to the brain. There is a significant evidence in the literature that drugs can be delivered directly from the nose to brain.21,22 Therefore, the IN route was chosen in the present study to promote drug distribution into the brain. Better brain targeting can be done by IN delivery, as it facilitates direct drug transport to the brain through the nasal olfactory and trigeminal nerve pathways, by passing BBB, avoiding gastrointestinal inactivation and first pass metabolism found in some oral drugs, allowing rapid drug absorption with a faster onset of action, increasing drug concentration and bioavailability in the brain, and therefore reducing the drug dose, and systemic side effects.21 Despite these advantages, the IN route has some limitations including: short residence time of the administered formulation due to mucociliary clearance, drug degradation by enzymes in the nasal cavity that impair drug delivery to the brain and limit its bioavailability, and limited dosing in terms of volume (50–200 μL) administered in the nasal cavity.23 Many recent literatures, less than five years, have been published on nose to brain delivery using nanoparticles, to overcome the IN limitations, such as nanoemulsion,24 polymeric nanoparticles,25 liposomes,26 solid lipid nanoparticles (SLNs)27 or nanostructured lipid carriers (NLCs),28 which are effective nose-to-brain drug delivery systems that improve the nasal permeability and bioavailability of drugs for effective brain targeting by increasing drug solubility and permeation, reducing mucociliary clearance and enzymatic degradation.29
In particular, formulations based on nanostructured lipid carriers (NLCs) are interesting candidates for nose to brain drug delivery without any modification to the drug molecule due to their rapid uptake by the brain, bioacceptability, biodegradability and safety.30 NLCs have been proposed as a second-generation of lipid nanoparticles to overcome the drawbacks of solid lipid nanoparticles (SLNs) including low drug loading efficiency and the possibility of drug expulsion during storage owing to the crystalline structure of SLNs. The matrix of NLCs is composed of a blend of solid and liquid lipids with different spatial arrangements; the incorporation of liquid lipid to solid lipid produces NLCs with less organized crystalline structure, resulting in greater imperfections in the crystal lattice to accommodate more drug molecules than SLNs, leading to improved drug loading and minimized drug expulsion.31 Generally, the particle diameter ranges from 10 to 1000 nm of NLC. NLCs offer prolongation of drug release and higher stability compared to liposomes and nanoemulsions, and have less toxicity than polymeric nanoparticles.32 NLCs have distinct advantages for nose-to-delivery; as they can boost the nasomucosal permeability of drugs due to their lipophilic nature, flexibility, and nano-size, the encapsulation of drug in the lipid matrix protects it against chemical and enzymatic degradation, delay drug elimination by mucociliary clearance and improve nasal retention time, allowing drug to reach directly to the brain at therapeutic level. They have rapid onset of action, longer duration and higher brain levels as compared to that of solutions. NLCs can also be loaded into hydrogel systems or coated with mucoadhesive polymers to improve these effects, and they do not cause nasal irritation or damage, owing to their natural lipid-based composition.33,34 Many formulations based on NLCs were developed for effective nose-to-brain drug delivery.35–37 Previous studies investigated the IN delivery of AST as SLNs38 or NLCs39 with the intention of improving brain targeting of AST for neurological disorders. Bhatt et al38 indicated, from biodistribution studies and gamma scintigraphy analysis, that higher AST concentration in the rat brain was achieved by IN administration of AST-SLNs, radiolabeled with technetium-99m (99mTc), as compared to the intravenous route. Pharmacokinetic studies of Gautam et al39 showed better availability of AST in the brain on the rats treated with AST-NLC in-situ gel (IN) as compared to those treated with AST-in-situ gel (IN).39
In view of this, this study aimed to prepare, optimize, and evaluate AST–loaded NLCs, for enhancing nose-to-brain delivery to improve its therapeutic efficacy. The optimized AST–NLC formulation was characterized in terms of particle size, polydispersity index, zeta potential, entrapment efficiency, drug loading, in vitro drug release, transmission electron microscopy, Fourier transform infrared spectroscopy, differential scanning calorimetry, and stability. We in vivo evaluated the therapeutic effects of the optimized formulation in AD-like rats after IN administration for 30 days, by pharmacodynamics studies, through measuring the levels of various biochemical parameters in the brain of rats such as; malondialdehyde, glutathione, nuclear factor-kappa B, caspase-3, acetylcholine, acetylcholinesterase, amyloid beta (Aβ1‑42), and β-site amyloid precursor protein cleaving enzyme-1 expression, and we explored the possible neuroprotective mechanisms of action of AST. Histopathological examination of the cortex and hippocampus was carried out.
Materials and Methods
Materials
Astaxanthin was purchased from Carbosynth Limited (Berkshire, UK). Glyceryl palmitostearate (Precirol® ATO 5) was obtained as a gift sample from Gattefossé (Saint-Priest Cedex, France). Poloxamer 188 (Pluronic® F68) was purchased from BASF SE (Ludwigshafen, Germany). Polysorbate 80 (Tween® 80) and oleic acid were purchased from Lobachemie laboratory reagents and fine chemicals (Mumbai, India). Methanol and acetonitrile HPLC grades were purchased from Sigma Aldrich (Germany). Phosphoric acid and sodium dihydrogen phosphate were purchased from El-Nasr Pharmaceutical Chemicals Co. (Cairo, Egypt).
Methods
Preparation of AST– Loaded NLCs
Different formulations of AST–NLCs were prepared using hot high-pressure homogenization (HPH) technique.40 Briefly, the solid lipid (glyceryl palmitostearate) was melted 5 ○C above its melting point, and then the liquid lipid (oleic acid) was added. Accurately weighed amount of AST was mixed in the above lipid melt. The aqueous phase was prepared by dissolving a surfactant mixture (consisted of poloxamer 188 and polysorbate 80, 50:50, %w/w) in 10 mL deionized water and heated to the same temperature as that of the above melted lipid phase. The hot aqueous surfactant solution was added dropwise to the melted lipid phase under continuous magnetic stirring (Magnetic stirrer IKA Labortechnik, Staufen, Germany) at 1000 rpm for 15 min to form a coarse pre-emulsion that was homogenized using high shear homogenizer (IKA T25 digital ultra-TURRAX, Staufen, Germany) at 15,000 rpm for 15 min. The formed nanoemulsion was cooled down to room temperature and stored at 4–8°C overnight to allow recrystallization of the lipid for NLCs formation.41 Processing variables such as total lipid-to-drug (L:D) ratio (ranging from 2:1 to 6:1,w/w), solid lipid-to-liquid lipid (SL:LL) ratio (ranging from 90:10 to 60:40, %w/w of total lipid amount), and concentration of surfactant mixture (ranging from 0.5 to 2.5%w/v) were tested and optimized to obtain optimal AST-NLC formulation (as shown in Table 1).42
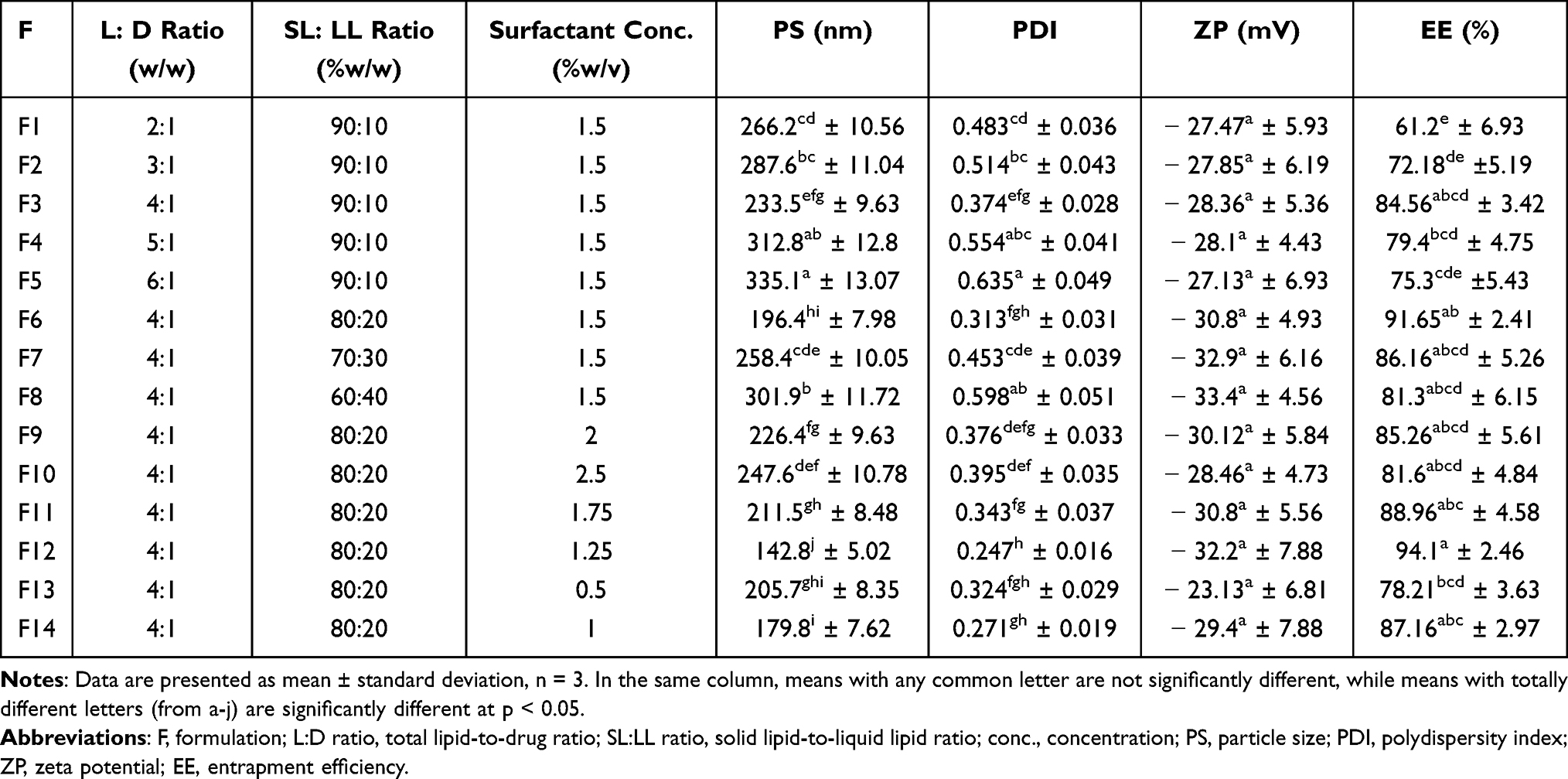 |
Table 1 Formulation Parameters and Characterization of Different AST–NLC Formulations |
In vitro Characterization of the Prepared AST–NLCs
Particle Size (PS), Polydispersity Index (PDI), and Zeta Potential (ZP)
The mean PS, PDI, and ZP of the prepared AST–NLCs were measured based on dynamic light scattering (DLS) method, also known as photon correlation spectroscopy (PCS) using Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). The dispersant (water) refractive index used for DLS measurement was 1.33 and the medium viscosity was 0.8872 cP. The material (lipid) refractive index was 1.40 and material absorbance was set at 0.01. The measurement using PCS is based on the light-scattering phenomena in which the statistical intensity fluctuations of the scattered light from the dispersed particles in the measuring cells are measured. An estimate for the mean size of the lipid particles, the intensity weighted mean diameter (often called z-average diameter or hydrodynamic diameter) and PDI were determined. ZP was measured by determining the electrophoretic mobility using the Malvern Zetasizer. Samples were diluted appropriately with filtered deionized water 10 times (1:10, v/v) prior to measurements to obtain suitable scattering intensity. Measurements were performed in triplicate at 25°C at a scattering angle of 173°, and results were reported as mean ± standard deviation (SD).43
Entrapment Efficiency (%EE) and Drug Loading (%DL)
%EE of the developed AST–NLCs was determined by an indirect method, depending on the separation of the aqueous phase containing free drug from drug-loaded NLCs by centrifugation. 5 mL of the formulation was centrifuged at 20,000 rpm for 30 min at 4°C using cooling ultracentrifuge (Sigma laborzentrifuge GmbH, Osterode, Germany). The supernatant containing free AST was analyzed by high performance liquid chromatography (HPLC, Agilent Technologies 1200 Infinity Series, USA) at λmax of 480 nm on a reversed phase C18 column (250 mm×4.6 mm, 5 μm) at room temperature, using an isocratic mobile phase consisted of acetonitrile and methanol (80:20,%v/v) with a flow rate of 1 mL/min, and the sample injection volume was 30 μL.
Standard solution of AST in methanol (100 µg/mL) was prepared and then diluted by the mobile phase solvents to obtain a series of AST solutions with known concentrations in the concentration range 0.39–100 μg/mL. The calibration curve was constructed by plotting peak areas against AST concentrations. A regression equation was obtained, and the coefficient of determination (R2) value was calculated. Unknown AST concentrations were calculated using the regression equation obtained. Measurements were done in triplicate. The method was validated for linearity, the limit of detection (LOD), limit of quantitation (LOQ) and specificity.44,45
%EE and %DL were calculated using the following equations:
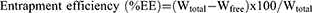

Where; Wtotal is the weight of total drug used, Wfree is the weight of free drug in the supernatant, and WLipids is the weight of lipids used in AST-NLCs.45,46
Transmission Electron Microscopy (TEM)
The optimized AST–NLC formulation was examined morphologically by transmission electron microscope (TEM, model JEM-100CX, JEOL, Japan). One drop of diluted samples was placed on a copper grid, stained with saturated solution of uranyl acetate, and air-dried at room temperature before analysis.43
Differential Scanning Calorimetry (DSC)
DSC was conducted to determine the thermal characteristics and crystalline state of pure AST, solid lipid, and AST–NLCs, using differential scanning calorimeter (Thermal Analysis Instruments, SDT Q600, USA). The samples (10 mg) were placed, and hermetically sealed in aluminum pans and heated at a rate of 10°C/ min from 25°C to 350°C under inert nitrogen flow at 20 mL/min.47
Fourier Transform Infrared (FTIR) Spectroscopy
The compatibility between AST and lipids used in NLCs was identified by Fourier transform infrared (FTIR) spectrophotometer (Perkin Elmer instruments, California, USA). FTIR spectra of pure drug, lipids, and AST–NLCs were scanned from 4000 cm−1 to 400 cm−1 and recorded.48
In vitro Drug Release and Release Kinetics Study
A dialysis bag diffusion technique was used to investigate the in vitro release of AST from the optimized AST–NLC formulation.49 Drug solution (10 mL) (in methanol/phosphate buffer pH 7.4) containing the same amount of AST (40 mg) in AST-NLCs (10 mL) was prepared as a control. 2 mL of AST–NLCs and AST–solution was placed into separate dialysis bags (VISKING molecular weight cutoff 12–14 KDa, London, UK), presoaked overnight in the release medium. The sealed bags were suspended in 100 mL of phosphate buffer (pH 7.4) and maintained at 37 ± 0.5°C in thermostatically controlled shaking water bath (Kottermann, type 3047, Hanigsen, Germany), at 100 rpm. Samples (5 mL) were withdrawn at designated time intervals up to 24 h, replaced with an equal volume of fresh medium to maintain sink condition, and analyzed by HPLC at λmax of 480 nm to determine the amount of AST released. Percent cumulative drug released versus time was plotted to illustrate the drug release pattern. Comparison between release profile of AST–NLCs and AST–solution was made by simple Student’s t-test at p < 0.05. In vitro release data of AST–NLCs were analyzed by various kinetic models such as zero order, first order, Higuchi, Hixon-Crowell, and Korsmeyer-Peppas models.50
Stability Study
The optimized AST-NLC formulation was stored at 4–8 ±2°C (refrigerator) and 25±2°C/60 ± 5% RH for six months. Samples were withdrawn after a period of 0, 1, 3 and 6 months and the effect on PS, PDI, and %EE was determined.49,51
In vivo Evaluation of the Optimized AST–NLC Formulation
Animals
The in vivo study was conducted on 30 male albino rats (150–200 gm) three months old purchased from the animal house of Medical Research Institute, Alexandria, Egypt. All rats had free access to food and water with 12:12 h light/dark cycle and constant environmental conditions before experimentation and thereafter. All procedures were performed in accordance with the Institutional Animal Care And Use Committee (IACUC)-Alexandria University, Egypt (Approval number: AU062019518251).
Induction of Alzheimer’s Disease
AD-like model was induced in rats using hydrated aluminum chloride (AlCl3.6H2O) solution that was given orally at a dose of 75 mg/kg body weight daily for 6 weeks.52 The Morris water maze (MWM) test was carried out at the end of the 6 weeks to evaluate the spatial memory and confirm the cognitive impairments in the rats.53
Experimental Design
Animals were divided into two main groups: Group I (Control group): consisted of 6 healthy male rats, and Group II (AD-like group): consisted of 24 AD-like rats that subdivided into 4 subgroups (6 rats each): Group IIA: untreated AD-like rats, Groups IIB, IIC, and IID: AD-like rats that received, by IN route, 20 µL (10 µL in each nostril) of AST–Solution (equivalent to 0.4 mg/Kg body weight), the optimized AST–NLC formulation (equivalent to 0.4 mg/Kg body weight), and free NLCs, respectively, daily for 30 days.38
After the last day of treatment, animals were subjected to the behavioral MWM test, then sacrificed by cervical dislocation under deep anesthesia by isoflurane and the brain was dissected. The cortex and hippocampus were excised from one hemisphere for tissue homogenate preparation and total RNA extraction for quantitative real-time polymerase chain reaction (qPCR). The other hemisphere was used for histopathological observation.
Tissue Homogenate Preparation
The excised cortex and hippocampus tissues were rinsed with saline and then homogenized in phosphate buffer saline (PBS) pH 7.4 in the ratio of 1:9. The homogenates were centrifuged at 10,000 rpm for 20 min at 4°C. The supernatants were divided into aliquots and stored at −20°C for subsequent determination of different biochemical parameters; amyloid beta (Aβ1-42), acetylcholine, acetylcholinesterase, reduced glutathione, malondialdehyde, nuclear factor-kappa B, caspase-3, and β-site amyloid precursor protein cleaving enzyme-1 expression.
Behavioral Test: Morris Water Maze (MWM) Test
Morris water maze (MWM) test was used to evaluate the spatial memory function in rats after AlCl3 induction and after the last day of IN treatments.53 The water maze consisted of a dark circular pool, 100 cm in diameter and 55 cm in height, filled with water (about 22 ± 2°C) to a depth of 20 cm, and made opaque by the addition of powdered starch. A submerged circular black platform (10 cm in diameter) was placed 20 cm away from the edge in a fixed location and 1 cm below the water surface. The pool was divided into 4 quadrants: north, south, east, and west (N-S-E-W) and several cues were placed outside the maze in a fixed position relative to the pool to help the rat locate the position of the escape platform hidden below the water surface. Each rat was placed in the water facing the wall of the pool and allowed to swim and find the hidden platform located in the northeast (NE) quadrant (target quadrant) of the maze. All rats were trained in the water maze for 4 consecutive days and the single training session consisted of four trials. During each trial, each rat was given 120 s to find the hidden platform. After mounting the platform, the animals were allowed to remain there for 20s, and were then placed in a holding cage for 30s until the start of the next trial. The time required for reaching the hidden platform (latency) was measured manually by using a stopwatch. After completion of training, the animals returned to their home cages.
On the fourth day, a probe trial (retention testing) was done which consisted of 60s of a free-swimming period for each rat with the hidden platform removed from the pool. The time spent in the target quadrant (expressed as a percent of the time spent in the pool) was calculated.
Biochemical Assays
Assays of Rat Amyloid Beta (Aβ1-42), Acetylcholine (ACh), Acetylcholinesterase (AChE), and Nuclear Factor-Kappa B (NF-κB; a Marker for Neuroinflammation) Contents
The cortical and hippocampal contents of Aβ1-42, ACh, AChE, and NF-κB were assayed according to the manufacturer’s instructions using Aβ1-42, ACh, AChE, and NF-κB rat ELISA kits, respectively, from Chongqing Biospes Co., Ltd., China.
Determination of Reduced Glutathione (GSH)
The enzymatic method described by Griffith et al54 was used to measure the reduced glutathione (GSH) content in the cortical and hippocampal tissues. This is a sensitive and specific enzymatic method that depends on the oxidation of GSH by 5,5ˋ-dithiobis-(2-nitrobenzoic acid) (DTNB) to yield GSSG and 5-thio-2-nitrobenzoic acid (TNB). Oxidized GSSG is reduced enzymatically by the action of glutathione reductase and Nicotinamide adenine dinucleotide phosphate (NADPH) to regenerate GSH that reacts again. The rate of TNB formation is monitored at 412 nm and is proportional with the GSH content of the sample.
Determination of Malondialdehyde (MDA, a Marker for Lipid Peroxidation)
Malondialdehyde (MDA) in the whole homogenate was determined according to the colorimetric method of Draper and Hadley.55 Briefly, the sample under test is heated with thiobarbituric acid (TBA) at low pH and the resulting pink chromogen has a maximal absorbance at 532 nm and proportional with the MDA content of the sample.
Determination of Caspase-3 Activity (a Marker for Apoptotic Cell Death)
Caspase-3 has a specificity for cleavage at the terminal side of aspartate residue of the amino acid sequence DEVD (Asp-Glu-Val-Asp). The caspase-3 enzymatic activity was assayed using caspase-3 assay Kit (Elabscainces, USA). This kit is used to conjugate caspase-3 sequence-specific peptides acetyl-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-pNA) to yellow groups p-nitroaniline (pNA). When the substrate is cleaved by caspase-3, the yellow group pNA is dissociated, which can be quantitated spectrophotometrically at 405 nm. The level of caspase enzymatic activity in the tissue homogenate is directly proportional to the color reaction. The final caspase-3 activity was normalized relative to the total protein content of each sample.
Gene Expression Analysis
Expression of β-Site Amyloid Precursor Protein Cleaving Enzyme-1 (BACE-1) Gene by Quantitative Real Time-Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from the cortical and hippocampal tissues using RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions, and the concentration and integrity of extracted RNA were checked using nanodrop. Reverse transcription was performed using miScript II RT Kit (Qiagen, Germany) according to the manufacturer’s instructions. The tissue expression of BACE-1 was quantified in the obtained cDNA using Rotor Gene SYBR Green PCR Kit (Qiagen, USA). The PCR amplification conditions were adjusted as an initial denaturation at 95℃ for 10 min and then 45 cycles of PCR for amplification as follows: denaturation at 95℃ for 20s, annealing at 55℃ for 20s, and extension at 70℃ for 15s. The relative expression of BACE-1 was quantified relative to the expression of the reference gene (18S rRNA) in the same sample by calculating and normalizing the threshold cycles (Ct) values of target BACE-1 to that of 18S using 2−ΔΔCt method.56 The primers used for the determination of rat genes are: BACE-1 (NM_019204.2) forward: 5’-GCATGATCATTGGTGGTATC-3’, and reverse: 5’-CCATCTTGAGATCTTGACCA-3’, and 18S rRNA (NR_046237.2) forward: 5’-GTAACCCGTTGAACCCCATT-3’, and reverse: 5’-AAGCTTATGACCCGCACTT-3’.
Histopathological Examination
Sample Preparation
The hemispheres were fixed with 10% para-formaldehyde and embedded in paraffin for making brain hemisphere blocks. Then, serial sections of the blocks were cut at 5 μm thick in a rotary microtome for hematoxylin and eosin (H and E) staining.
Hematoxylin and Eosin (H and E) Staining
In brief, after the paraffin sections were dewaxed, hematoxylin staining was performed for 3 s, followed by eosin staining for 3 min, and then the sections were dehydrated with alcohol, cleared with xylene, and sealed. The histopathological lesions were investigated under a light microscope and photographed using a digital camera (Nikon Corporation Co., Ltd., Japan).57
Statistical Analysis
Data were analyzed using IBM SPSS software package version 20.0 (Armonk, NY: IBM Corp). The data were expressed as mean ± standard deviation (SD) and analyzed using one-way analysis of variance (ANOVA) followed by Tukey multiple comparison post-hoc tests to compare between different groups and Pearson for correlation study. The P-value was assumed to be significant at p < 0.05.
Results and Discussion
Preparation and Optimization of AST-NLCs
Different AST-NLC formulations were prepared using hot high-pressure homogenization (HPH) technique, which is simple, quick at laboratory scale and there is no use of organic solvents during the development of the formulation.58
Different processing variables such as total lipid-to-drug (L:D) ratio, solid lipid-to-liquid lipid (SL:LL) ratio, and concentration of surfactant mixture were tested and optimized, as they affect the physicochemical properties of the prepared formulations such as the mean particle size (PS), polydispersity index (PDI), zeta potential (ZP), and entrapment efficiency (%EE).58,59 From the results obtained in Table 1, the average PS of the prepared formulations of AST-NLCs ranged from 142.8±5.02 nm to 335.1±13.07 nm, and PDI ranged from 0.247±0.016 to 0.635± 0.049. All formulations exhibited negative ZP, which was in the range of –23.13± 6.81 to –33.4±4.56 mV, and EE was in the range of 61.2± 6.93 to 94.1±2.46% due to the variation in formulation parameters.
Effect of Formulation Variables on PS, PDI, and ZP of AST-NLCs
From the results obtained in Table 1, it was observed that the mean PS and PDI of the prepared AST-NLCs significantly decreased with increasing total lipid amount relative to drug (L:D ratio) from 3:1 (F2) to 4:1,w/w (F3) and significantly increased with further increase of L:D ratio to 5:1 (F4) or 6:1,w/w (F5). This can be explained in terms of tendency of lipid to coalesce at high lipid concentration, due to the decreased surfactant emulsifying efficiency to coat all lipid droplets and increase dispersion viscosity that result into higher surface tension, aggregation of particles and thus higher PS and PDI.60 At a constant L:D ratio, an initial increase in the amount of liquid lipid to 20% with respect to solid lipid, changing solid lipid-to-liquid lipid (SL:LL) ratio, from 90:10 (F3) to 80:20, %w/w (F6) significantly decreased PS and decreased PDI but not significantly. At SL:LL ratio of 80:20,%w/w, there is a perfect blend of solid and liquid lipids and no oil expulsion from the lipid blend was detected. Liquid lipid can decrease the viscosity and surface tension of the system that offers NLCs with higher surface area, smaller PS and PDI. Further increase in the amount of liquid lipid more than 20% with respect to solid lipid, changing SL:LL ratio from 80:20 (F6) to 70:30 (F7) or 60:40, %w/w, (F8), significantly increased PS and PDI. Higher liquid lipid amount could facilitate lipid coalescence and increase the size of the produced vesicles through swelling and disruption of the NLC wall that may result in increased NLC size, and the solid and liquid lipids will not to be well mixable.61 At constant L:D and SL:LL ratios, an initial increase of surfactant concentration from 0.5%w/v (F13) to 1.25%w/v (F12) significantly decreased PS and increased ZP but not significantly, as it was sufficient to reduce the interfacial tension, cover the surface of nanoparticles effectively, and prevented their agglomeration. Once the surfactant concentration reaches saturation point, further increase in surfactant concentration above 1.25%w/v (F6, F11, F9 and F10), significantly increased PS and decreased ZP but not significantly, due to deposition of excess surfactant molecules on the NLC surface and agglomeration of particles. The negative charge in all formulations is attributed to their lipid composition due to the presence of free fatty acids at the surface of the particles. Primary emulsion was better stabilized by the surfactant mixture of poloxamer 188 and polysorbate 80, being safe, biodegradable and non-ionic surfactants with good emulsification efficacy that stabilize the system by steric stabilization that does not allow particles to come closer to each other and thus particle agglomeration is prevented.62,63
Effect of Formulation Variables on %EE of AST-NLCs
It was observed that %EE of the prepared formulations significantly increased with increasing L:D ratio from 2:1 (F1) to 4:1,w/w (F3), due to solubilization of the lipophilic drug in the lipid and reduced drug escaping into the external phase. Further increase in L:D ratio (F4 & F5) promoted the expulsion of already entrapped drug and decreased EE.62 An initial increase in the amount of liquid lipid to 20% with respect to solid lipid, changing SL:LL ratio from 90:10 (F3) to 80:20, %w/w (F6) increased %EE but not significantly. The addition of liquid lipid (oil) to the solid lipid in the NLC matrix causes distortion of crystalline structure of the lipid which is favourable for achieving higher EE and increase the amount of liquid lipid enhances the solubilization capacity of total lipid phase thus entrapping more drug during formulation. Further increase in the amount of liquid lipid more than 20% with respect to solid lipid, changing SL:LL ratio from 80:20 (F6) to 70:30 (F7) or 60:40, %w/w, (F8), decreased %EE. This may be due to lipid precipitation, which occurs during particle production. After NLC formulation, when it was cooled, recrystallization of lipids occurs resulting into a drug-free core or a core with reduced drug content.61 An initial increase of surfactant concentration from 0.5%w/v (F13) to 1.25%w/v (F12), significantly increased %EE, as the increase in surfactant concentration could enhance the drug partitioning and facilitate its solubilization in the lipid phase and stabilize the system. Once the lipid matrix get saturated, a further increase of surfactant concentration above 1.25%w/v (F6, F11, F9 and F10), a reduction of %EE was observed. This may be attributed to drug expulsion from the lipid matrix, due to entrapment of surfactant molecules into the NLCs at higher surfactant concentration and increased drug escaping into the external phase.64,65
From the results obtained, it could be suggested that formulation (F12) was selected as the optimal AST-NLC formulation, as it had a significant smaller mean PS than other formulations, lower PDI, the highest %EE and high ZP.
In the optimized formulation (F12); L:D ratio was 4:1 (w/w), the amount of AST = 40 mg, and total lipid amount = 160 mg; SL:LL ratio was 80:20 (%w/w total lipid amount), the amount of solid lipid = 128 mg (80% of total lipid amount) and liquid lipid amount = 32 (20% of total lipid amount), concentration of surfactant mixture was 1.25% w/v and volume of deionized water was 10 mL.
In vitro Characterization of the Optimized AST–NLC Formulation (F12) Particle Size (PS), Polydispersity Index (PDI) and Zeta Potential (ZP)
The mean PS is a crucial parameter for nanocarriers, as it has a significant impact on their biopharmaceutical characteristics including release pattern, absorption, and distribution in the biological system. The smaller size may lead to a more efficient permeation of the drug via the nasal mucosa directly to the brain, rapid absorption and improves drug distribution and bioavailability in the brain. It was reported that nanoparticles having average diameter up to 200 nm could be easily transported transcellularly via intranasal route directly to the brain.66,67 The optimized AST–NLC formulation (F12) had a mean PS of 142.8 ± 5.02 nm (less than 200 nm), which was found to be suitable for brain targeting through IN route. PDI is an indicator of the quality of dispersion, ensures the mono- or polydispersity of dispersions, and plays an important role to predict the chance of agglomeration. The smaller value of PDI is considered ideal for nanoparticle homogeneity and to ward off aggregation. Lower PDI value might be associated with a high homogeneity in the particle population, whereas high PDI values suggest a broad size distribution.67 PDI value of the optimized formulation was found to be 0.247 ± 0.016 (less than 0.3), indicating narrow particle size distribution. ZP characterizes the electrical charge on the NLC surface and gives an idea about the aggregation tendency and long-term physical stability of NLCs. A suitable high value of ZP (less than −30 mV or greater than +30 mV) confers stability because particles resist aggregation.63,68 The optimal formulation had high ZP (–32.2 ± 7.88 mV), indicated high stability.
Entrapment Efficiency (%EE) and Drug Loading (%DL)
%EE and %DL of the optimized formulation were 94.1 ± 2.46% and 23.5 ± 1.48%, respectively. The high %EE may be attributed to the incorporation of liquid lipid to solid lipid in the NLC matrix, resulting in crystal lattice defects, which provides enough space to accommodate more molecules of lipophilic drug (AST) in the matrix. It may be also due to the affinity and solubility of the lipophilic drug in the lipid nanoparticle matrix, the decreased solubility of the drug in the external phase, the small mean PS, the uniform size distribution, and stability of the prepared formulation that minimized drug leakage from nanoparticles.61,62
The concentration of free drug was determined using a simple, rapid, and validated HPLC method.45 Under the chromatographic conditions applied, sharp symmetric peak was obtained for AST with a retention time of 4.1 min. The resulting chromatogram showed no interfering peaks indicating specificity of the method. Linearity was checked in the concentration range 0.39 to 100 μg/mL by plotting peak areas against AST concentrations to obtain the calibration curve. The coefficient of determination (R2) was 0.9991, revealed a good linearity and indicating good correlation between peak area and drug concentration within the concentration range tested. The limits of detection (LOD) and quantitation (LOQ) were 0.26 μg/mL and 0.79 μg/mL, respectively.
Transmission Electron Microscopy (TEM)
The morphological study of the optimized AST-NLC formulation using TEM (Figure 1) revealed that the nanoparticles are nonaggregated and spherical in shape with a smooth surface and uniform particle size distribution. PS measured by TEM was smaller than that determined by PCS using Malvern Zetasizer, as PCS measured the hydrodynamic diameter, whereas TEM measured the true size of dried particles.43
 |
Figure 1 Transmission electron microscopy (TEM) photograph of the optimized AST–NLC formulation (40,000X magnification). |
Differential Scanning Calorimetry (DSC)
DSC was employed to investigate the melting and crystallization behavior of the solid lipid and drug. The DSC thermograms (Figure 2) showed that pure AST had a sharp melting endothermic peak at 221.14°C and a small broad endothermic peak at 273.78°C, while solid lipid (glyceryl palmitostearate) alone showed an endothermic peak at 62.75°C, indicating their crystalline nature. The melting peaks of AST completely disappeared in the DSC thermogram of AST-NLCs, indicating that the drug was molecularly dispersed in the NLC matrix and converted to an amorphous state, and pinpointing complete solubilization of AST inside the lipid matrix. There was a slight shift in the melting peak of glyceryl palmitostearate (from 62.75°C to 59.78°C), a reduction of the melting onset (from 55.42°C to 50.01°C) and enthalpy (from 191.1 J/g to 123.4 J/g) in AST-NLCs. This could be attributed to the presence liquid lipid, which creates distortion in the lipid matrix, resulting in less ordered crystals with decreased enthalpy.47,69
 |
Figure 2 Differential scanning calorimetry (DSC) thermograms of (a) glyceryl palmitostearate, (b) astaxanthin, and (c) astaxanthin-nanostructured lipid carriers (AST–NLCs). |
Fourier Transform Infrared (FTIR) Spectroscopy
FTIR was used to evaluate the molecular interaction between AST and the lipid matrix of NLCs. It was observed from FTIR spectra (Figure 3) that the characteristic peaks of pure AST were found in FTIR spectrum of AST–NLCs in the same position without any distinct shift and have an almost equal intensity, indicating no chemical interaction occurred between AST and lipids used in NLCs.48
 |
Figure 3 Fourier transform infrared (FTIR) spectra of (a) oleic acid, (b) glyceryl palmitostearate, (c) astaxanthin, and (d) astaxanthin-nanostructured lipid carriers (AST–NLCs). |
In vitro Release and Release Kinetics Study
The in vitro release of AST from the optimized AST–NLC formulation (Figure 4) demonstrated a biphasic release pattern with an initial burst release of 17.4% in the first hour, followed by slow and sustained release up to 24 h. The biphasic release behavior of NLCs might be due to the imperfect arrangement of the solid and liquid lipid blend of the NLC matrix. The drug embedded in the solid lipid crystals might provide slow and prolonged release, while the immediate release could be attributed to the drug present in the liquid lipid, as the drug diffusion through the liquid lipid phase was faster than that via the solid lipid phase. Some drug might also present in the outer shell as free moiety, which also contributed to the burst release in the first hour. The melted blend of solid and liquid lipids increased the viscosity of the particle matrix, leading to a slower drug release rate.34 On the other hand, AST–solution showed immediate release of 81.3% in the first hour and almost all the drug content (99.1%) released in 4 h. The later slow release from the solution might be due to the lack of concentration gradient between donor and receptor compartment, AST was present in solution form inside the dialysis bag. AST solution was used as a control to ensure dialyzability of the drug and to demonstrate the in vitro release difference between drug in solution form and drug entrapped in NLCs. The permeation of the drug across the dialysis membrane was the controlling step for the drug presence in the receiving compartment of the dialysis bag. This could be explained that the dialysis membrane might have hindered drug release. Also, the release depends on the diffusion coefficient of the drug.70
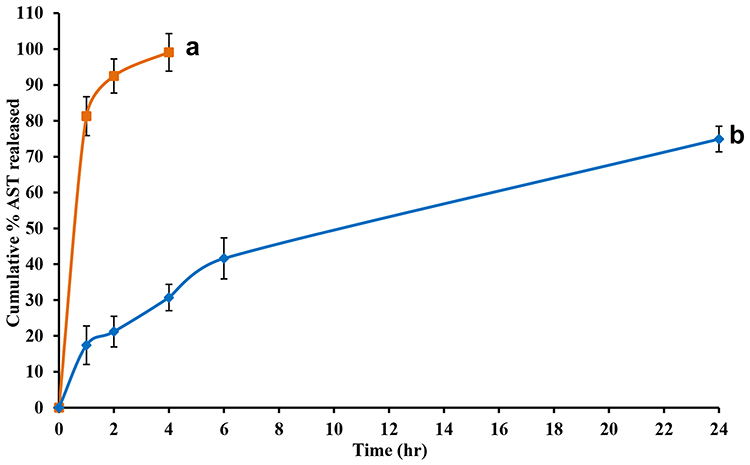 |
Figure 4 In vitro release profile of astaxanthin from (a) astaxanthin–solution (AST–Sol), and (b) astaxanthin-nanostructured lipid carriers (AST–NLCs) for 24 h using dialysis method in phosphate buffer (pH 7.4) at 37 ± 0.5°C. Data are presented as mean ± standard deviation, n = 3. |
A significant difference (p < 0.05) was marked between AST–solution and AST–NLCs after 4 hr. Korsmeyer-Peppas model was the best fitted model for AST release from NLCs, as it had the highest correlation coefficient (r2 = 0.9959) (Table 2), and the release mechanism involved diffusion and matrix erosion, as the value of release exponent “n” was 0.476 (between 0.45 and 0.89).71
 |
Table 2 In vitro Release Kinetics Models for the Optimized AST–NLC Formulation |
Stability Study
Slight non-significant change was observed in the mean PS, PDI, and %EE of AST-NLCs when they were stored at 4–8 ±2°C (refrigerator) for six months (Table 3). PS and PDI of AST-NLCs increased significantly after storage at 25±2°C/60 ± 5% RH for six months, as the kinetic energy of particles increases at higher temperature leads to particle aggregation, and %EE was reduced with time but not significantly. Therefore, storage of AST–NLCs was preferred to be at 4–8 ±2°C.
 |
Table 3 The Characteristic Parameters of AST–NLCs After a 6-Month Stability Study at Different Storage Conditions |
In vivo Evaluation of the Optimized AST–NLC Formulation
Behavioral Test: Morris Water Maze (MWM) Test
Latency Time
The results of the time required for reaching the hidden platform (latency) during 4 days of training trials before and after treatment are presented in Table 4. At the baseline (before treatment), on the first day, there was a significant difference between the normal control rats and all AD-like rats in the latency time to reach the hidden platform with no significant differences between the AD-like groups. As training proceeds, the control rats showed a decline in the latency time. However, the AD-like rats showed no significant difference during the 4 days of training trials. After treatment, as training proceeds, there was a significant difference in the latency time to reach the hidden platform between AD-like untreated and treated rats with AST-NLCs or AST-solution, and rats treated with AST-NLCs showed a significant decline in the latency. It was observed that there was no significant difference in the latency time between control rats and AD-like rats treated with AST-NLCs, but a significant difference in the latency time was observed between control rats and AD-like rats treated with AST-solution, during the 4 days of training trials. The AD-like rats treated with AST-NLCs showed significantly less latency time to reach the hidden platform compared to AST-solution on the 2nd and 4th days.
 |
Table 4 Morris Water Maze (MWM) Test During 4 Days of Training and Probe Trail Before and After Treatment |
Probe Trial
Before treatment, all AD-like rats showed a significant decline in the percentage of the time spent in the target quadrant of the pool in the probe trial as compared to the control rats, with no significant differences between the AD-like groups (Table 4). After treatment, AD-like rats treated with AST-NLCs or AST-solution showed a significant increase in the percentage of time spent in the target quadrant as compared to the control and untreated AD-like rats. The rats treated with AST-NLCs showed significantly higher percentage of time spent in the target quadrant than AST-solution.
In the present study, the non-genetic AD-like model was induced in rats by oral administration of AlCl3 for 6 weeks.52 This model mimics to large extent the pathophysiology of AD in humans including long-term exposure to aluminum (Al). Aluminum is neurotoxic even in small amounts, accumulates in the brain over a lifetime, and contributes to the development of AD. In the present study, the established AD-like rat model showed cognitive impairment and behavioral changes as indicated by the long latency period during the four days of MWM and probe trial experiments.
AST is a natural lipid-soluble xanthophyll carotenoid with neuroprotective potential due to its antioxidant, anti-inflammatory and anti-apoptotic properties, and it has the ability to cross the BBB.6 The results indicated that AD-like rats treated intranasally with the optimized AST-NLC formulation showed significantly better performance in the MWM than the untreated rats and rats treated with free drug solution, as NLCs could improve the nasal permeability and nose to brain drug delivery. Taksima et al10 indicated that rats with AD treated with AST decreased their escape latency time, increased the time spent in the target quadrant in the MWM test and decreased Aβ protein and neurodegeneration. Rahman et al72 found that AST reversed the cognitive and memory impairment, assessed by MWM test and Novel object Recognition test in Aβ infused rats, and significantly attenuated Aβ (1–42) level.
Biochemical Assays
Amyloidogenic Pathway
Amyloid Beta (Aβ1-42) Content and β-Site Amyloid Precursor Protein Cleaving Enzyme-1 (BACE-1) Expression
The AD-like untreated rats showed significantly higher Aβ1-42 content than control rats by about 3.5-fold in both cortex and hippocampus (Figure 5), and a significant upregulation of BACE-1 expression compared to control rats by about 1.37-fold and 6.5-fold in the cortex and hippocampus, respectively (Figure 6); the upregulation of BACE-1 expression in the hippocampus was higher than the cortex. The rats treated intranasally with AST in NLCs or solution showed significantly lower cortical and hippocampal Aβ1-42 content and BACE-1 expression than the untreated rats. The rats treated with AST-NLCs had significantly lower cortical and hippocampal Aβ1-42 content and lower hippocampal BACE-1 expression compared with drug solution, while there is no significant difference in the cortical BACE-1 expression between AST-solution and AST-NLCs. Treatment with AST-NLCs completely normalized the cortical Aβ1-42 content and BACE-1 expression, while in the hippocampus, they are still significantly higher than control rats.
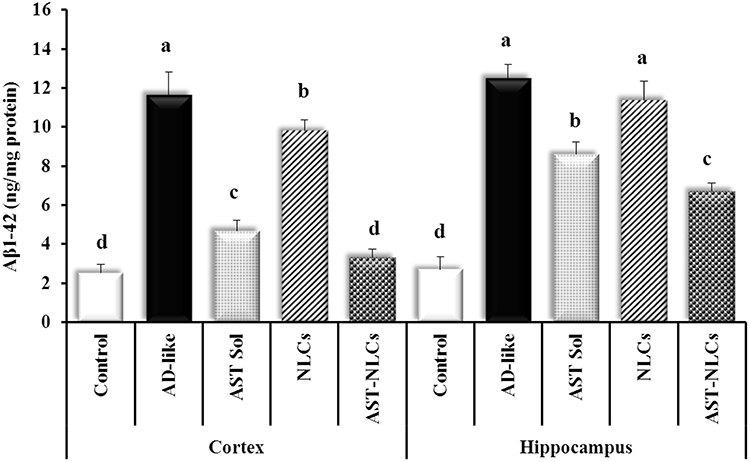 |
Figure 5 Amyloid beta (Aβ1-42) content in the cortex and hippocampus of the studied groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (cortex or hippocampus), means with any common letter are not significantly different, while means with totally different letters (from a – d) are significantly different at p < 0.05. Abbreviations: AD-like, Alzheimer’s disease-like; AST, astaxanthin; Sol, solution; NLCs, nanostructured lipid carriers. |
 |
Figure 6 β-site amyloid precursor protein cleaving enzyme-1 (BACE-1) expression in the cortex and hippocampus of the studied groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (cortex or hippocampus), means with any common letter are not significantly different, while means with totally different letters (from a-d) are significantly different at p < 0.05. Abbreviations: AD-like, Alzheimer’s disease-like; AST, astaxanthin; Sol, solution; NLCs, nanostructured lipid carriers. |
In the present study, AlCl3 induced neurochemical alterations in the cortex and hippocampus of AD-like rats.52 Regarding the amyloidogenic pathway, the AD-like rats showed significant upregulation in BACE-1 gene expression and have a marked increase in Aβ1-42 contents in both the cerebral cortex and hippocampus, which may confirm the metabolic shifting toward β-degradation of amyloid precursor protein (APP). Aβ1-42 aggregates (senile plaques) cause cellular damage in the brain, as they have been shown to contribute to the formation of neurofibrillary tangles (NFTs), oxidative damage, excitotoxicity, neuro-inflammation, and cell death. BACE-1 is the major β-secretase present in the brain and considered to be the rate-limiting step in the production of neurotoxic Aβ. Since BACE-1 catalyzes the cleavage of APP to produce the soluble APP and the membrane-bound C-terminal fragment (C99), it plays a significant role in the amyloidogenic pathway. The latter peptide is then cleaved by γ-secretase to produce the neurotoxic amyloid peptide fragment (Aβ1-42), which can aggregate and form the senile plaques, which are one of the hallmarks of AD. Therefore, inhibitors of BACE1, which is seen as a key therapeutic target, may be used to treat AD.73,74 The results clearly indicated the potential anti-amyloidogenic effect of AST especially in NLCs through downregulating BACE-1 expression and subsequent inhibition of Aβ1-42 production.
Previous studies have demonstrated the neuroprotective role of AST against Aβ oligomers both in vitro and in vivo. The probable reason for the anti-amyloidogenic action of AST can be due to its potent antioxidant effects and reduction of mitochondrial ROS production or maybe via significant reduction in BACE-1 and γ-secretase expression, which leads to the attenuation of Aβ production and formation of Aβ plaques.10,72 Elham et al75 found that AST downregulated transcription and activity of amyloidogenic BACE-1, reduced Aβ oligomers, enhanced Aβ clearance, and upregulated the disintegrin and metalloproteinase domain-containing protein 10 (ADAM 10) and α-secretase, responsible for the non-amyloidogenic processing of APP, and increased soluble APPα production.76
Cholinergic Pathway
Acetylcholine (ACh) Content and Acetylcholinesterase (AChE) Activity
The AD-like untreated rats showed significantly lower ACh content (by 57.83% and 64.35%), and significantly higher AChE activity (by 146.8% and 122.7%) than control rats in the cortex and hippocampus, respectively (Table 5). Treatment with AST in NLCs or solution showed a significant increase in the cortical and hippocampal ACh level compared to the untreated rats. The rats treated with AST-solution showed no significant difference in the cortical AChE activity compared to the untreated rats. In the cortex, no significant difference was observed in ACh level between AST in NLCs or solution, while AST-NLCs showed a significant decline in AChE activity compared with AST-solution. In the hippocampus, the rats treated with AST-NLCs had significantly higher ACh content and a significant decline in AChE activity compared to AST-solution.
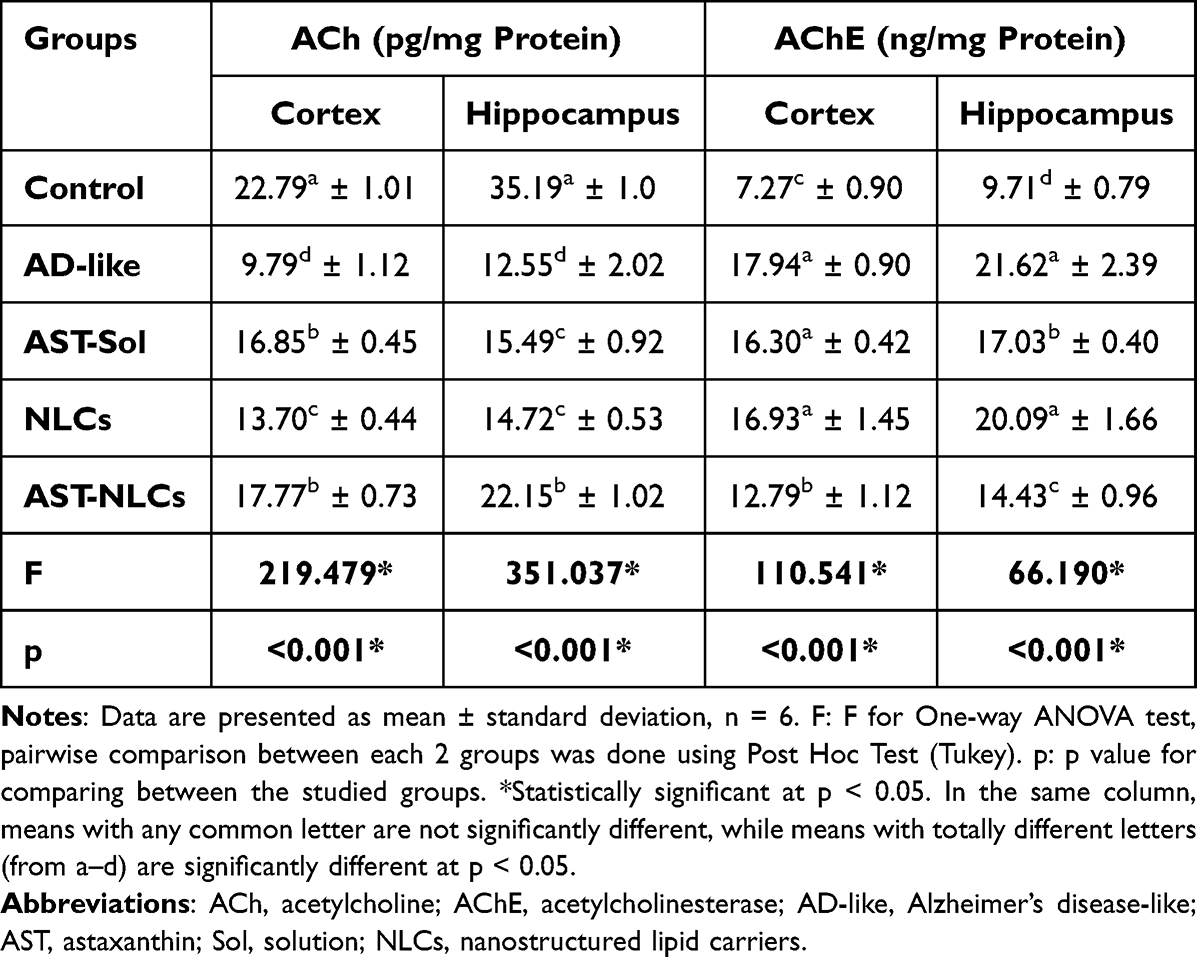 |
Table 5 Acetylcholine (ACh) Content and Acetylcholinesterase (AChE) Activity in the Cortex and Hippocampus of the Studied Groups |
Aβ1-42 had direct and indirect deleterious effects on neurotransmission, organelle function, signaling cascades, axonal transport, and immunological response that result in synaptic loss and dysfunctions in neurotransmitter release such as acetylcholine (ACh), which is fundamental in many aspects of cognition.73 The cholinergic pathway is affected in AD-like rats as the cerebral cortex and hippocampus showed significantly lower ACh level and higher AChE activity compared to control rats. These results are in line with Hafez et al77 and Yassin et al,78 who found decreased ACh levels in rats with AlCl3-induced AD. ACh is one of the brain’s main excitatory neurotransmitters. AChE activity responsible for the hydrolysis of ACh to terminate its excitatory action and the increased AChE activity in AD may be the cause for rapid destruction of ACh and declining its levels.79 A 2-day exposure to Aβ1-42 decreased the intracellular ACh concentration in the cholinergic hybrid SN56 cell line, suggesting that the rise in Aβ1-42 levels may be responsible for the fall in ACh levels.80 The suppression of the amyloidogenic pathway in AD-like rats treated with AST-NLCs was associated with significant correction of ACh and AChE contents in both cortex and hippocampus, indicating the anti-acetylcholinesterase potential of AST. Rahman et al72 found that AST significantly attenuated AChE level in the rat hippocampus and reversed the cognitive and memory impairment.
Redox Parameters
Reduced Glutathione (GSH) and Malondialdehyde (MDA) Contents
The AD-like untreated rats showed significantly lower GSH level (by 29.53% and 41.5%), and significantly higher MDA level (by 166.97% and 95.83%) than control rats in the cortex and hippocampus, respectively (Table 6). The rats treated with AST in NLCs or solution showed a significant elevation of the GSH level and a significant decline in the MDA level in both cortex and hippocampus compared to the untreated rats. In the cortex, the rats treated with AST-NLCs showed a significant decline in the MDA level and non-significant difference in the GSH level compared to AST-solution. In the hippocampus, AST-NLCs showed a significant increase in the GSH level and a significant decline in the MDA level compared to AST-solution. Treatment with AST-NLCs completely normalized the cortical GSH and the hippocampal MDA levels, while the hippocampal GSH level is still significantly lower and the cortical MDA level is still significantly higher than control rats.
 |
Table 6 Reduced Glutathione (GSH) and Malondialdehyde (MDA) Contents in the Cortex and Hippocampus of the Studied Groups |
Patients with AD showed a significant degree of oxidative damage in the brain.5 The brain is a target for oxidative stress due to the presence of high levels of unsaturated fatty acids. In AD, overproduction of reactive oxygen species (ROS) can result in protein and lipid oxidation, DNA and cell damage, and mitochondrial dysfunction, that drive the formation of additional Aβ1-42, which ultimately leads to neuron death.81 Considering the role of oxidative stress in neurodegenerative disorders, there is a growing interest in antioxidant therapies. Astaxanthin is a natural potent antioxidant compared to β-carotene and Vitamin. AST has a polar region (hydroxyl and ketone moieties) at each end of the molecule’s ionone rings and central nonpolar zone made up of 13 conjugated double bonds (polyene chain) that enable AST to scavenge and quench ROS, terminate free radical chain reactions by either donating or accepting electrons, and without being destroyed or becoming a pro-oxidant in the process, and inhibiting lipid peroxidation within the cell membrane as well as at the surface, and prevent oxidative DNA damage.82
Previous researches conducted in animal models have also identified a potential for astaxanthin to indirectly upregulate the endogenous antioxidant defense system including; superoxide dismutase (SOD), catalase, glutathione peroxidase, thiobarbituric acid reactive substances (TBARS), heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase-1 (NQO-1), and glutathione-S-transferase-α1 (GST-α1), by increasing expression of redox sensitive transcription factors, such as nuclear factor erythroid 2-related factor 2 (Nrf2) through activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway and the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated protein kinase (ERK) pathway.83,84 Nrf2 activation promotes the re-establishment of balance between oxidants and endogenous antioxidants after oxidative insult. Also, the increase in Nrf2 level increases the expressions of genes implicated in repair/removal of damaged DNA/proteins that enhance cell survival and potently protects neuronal cultures from Aβ toxicity.85 Nakagawa et al86 found that AST supplementation resulted in improved erythrocyte antioxidant status, reduced phospholipid hydroperoxides (PLOOH) levels, that resulted from lipid peroxidation, and reduced oxidative DNA damage in N-methyl-D-aspartate (NMDA)-induced excitotoxicity model, suggesting that astaxanthin may contribute to the prevention of dementia.
Malondialdehyde (MDA) formed by free radicals during ionization events in the body, and the level of MDA in the brain increases during oxidative stress and it is used as a marker of lipid peroxidation. Reduced glutathione (GSH) is a vital endogenous protective antioxidant against oxidative stress, which is oxidized to oxidized glutathione (GSSG), the total GSH level and GSH/GSSG ratio are indices of the protective ability of cells under oxidative stress. In the present study, the oxidative stress in the brain of AD-like rats is indicated by the decline of the GSH level and marked elevation in the MDA level in both cortex and hippocampus.87 Treatment with AST, especially in NLCs, was associated with potent antioxidant potential as indicated by the significant increased GSH and decreased MDA levels. In accordance with the present work, Akkoyun et al88 found that AST administration attenuated elevated MDA level and increased lower GSH level and superoxide dismutase (SOD) activity in rat brain due to cadmium (Cd) administration.
Nuclear Factor-Kappa B (NF-κB): A Marker for Neuroinflammation
The AD-like untreated rats showed marked higher NF-κB level compared to control rats by 581.36% and 699.1% in the cortex and hippocampus, respectively (Figure 7). Treatment with AST in NLCs or solution significantly decreased the cortical and hippocampal NF-κB level compared with the untreated rats. There was non-significant difference in the cortical NF-κB level after treatment with AST in NLCs or solution, while AST-NLCs showed significantly lower hippocampal NF-κB level compared to AST-solution.
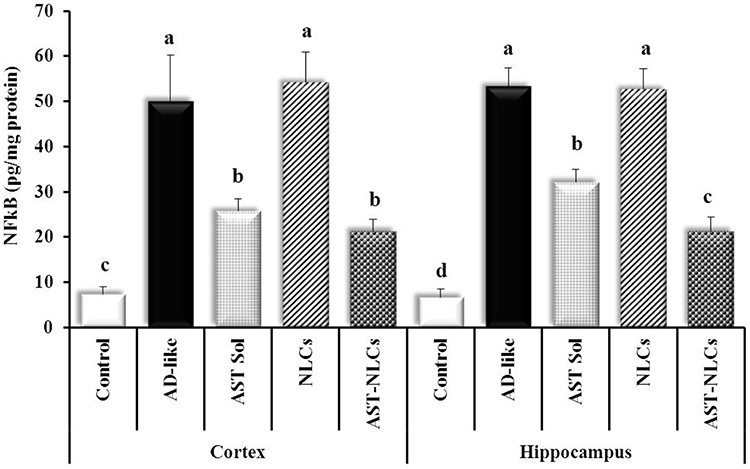 |
Figure 7 Nuclear factor-kappa B (NF-κB) content in the cortex and hippocampus of the studied groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (cortex or hippocampus), means with any common letter are not significantly different, while means with totally different letters (from a – d) are significantly different at p < 0.05. Abbreviations: AD-like, Alzheimer’s disease-like; AST, astaxanthin; Sol, solution; NLCs, nanostructured lipid carriers. |
In AD, oxidative stress and high Aβ level activate the body’s immune response, resulting in neuro-inflammation. The nuclear factor-kappa Beta (NF-κB) signaling pathway is an important and ubiquitous nuclear transcription pathway that serves important roles in inflammatory and immune responses. Excessive activation of the NF-κB signaling pathway is related to inflammatory changes in neurological disorders, like AD. In unstimulated conditions, NF-κB remains inactive in the cytoplasm and interacts with the inhibitory (IκB) family (IκB-α). In AD, NF-κB is activated through dissociation of IκB, which is phosphorylated by the IκB kinase complex (IKKα and IKKβ). Dissociated NF-κB enters the nucleus and binds to κB regulatory elements that produce the pro-inflammatory cytokines; interleukin-1 β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α). Therefore, blocking the NF-κB signaling pathway is important for the mediation of inflammatory diseases.89 Astaxanthin can block excessive NF-κB signaling by downregulating the phosphorylation of IκB-α or increasing the cellular expression of IκB-α mRNA and protein, and downregulate the expression of pro-inflammatory cytokines.90,91
In the present study, the induced oxidative stress in AD-like rats was associated with induction of neuroinflammation as indicated by marked elevation of the cortical and hippocampal NF-κB contents.92 Treatment of AD-like rats with AST-NLCs was associated with anti-neuroinflammatory potential as indicated by the decreased NF-κB content. Park et al93 reported astaxanthin reduced the DNA oxidative damage biomarker inflammation, thus enhancing immune response. Rahman et al72 found that AST significantly attenuated TNF-α level in the rat hippocampus. Kim et al,94 study in PM2.5-induced neuroinflammation model, demonstrated that AST treatment decreased the expression of M1 pro-inflammatory cytokines (IL-1β, TNF-α and IL-6) and increased the expression of M2 anti-inflammatory cytokines (IL-10 and arginase-1). AST also exhibits anti-inflammatory effects by inhibiting cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) through the regulation of multiple genes. Han et al95 investigated that AST administration ameliorated lipopolysaccharide (LPS)-induced memory loss, reduced LPS-induced expression of inflammatory proteins, production of ROS, nitric oxide (NO), cytokines and chemokines, and also reduced LPS-induced B-secretase and Aβ 1–42 generation through the down-regulation of amyloidogenic proteins both in vivo and in vitro. Furthermore, AST suppressed the DNA binding activities of the signal transducer and activator of transcription 3 (STAT3) that plays critical roles in inflammatory diseases, including AD. Che et al96 found that AST is effective in controlling oxidative stress, inflammasome production and activation, and reducing Tau hyper-phosphorylation.
Caspase-3 Activity: A Marker for Apoptotic Cell Death
The AD-like untreated rats showed significantly higher caspase-3 activity than control rats by 513.1% and 546.45% in the cortex and hippocampus, respectively (Figure 8). The rats treated with AST in NLCs or solution significantly decreased the cortical and hippocampal caspase-3 level compared to the untreated rats. AST-NLCs result in a significant decline in caspase-3 level compared to AST-solution in both cortex and hippocampus.
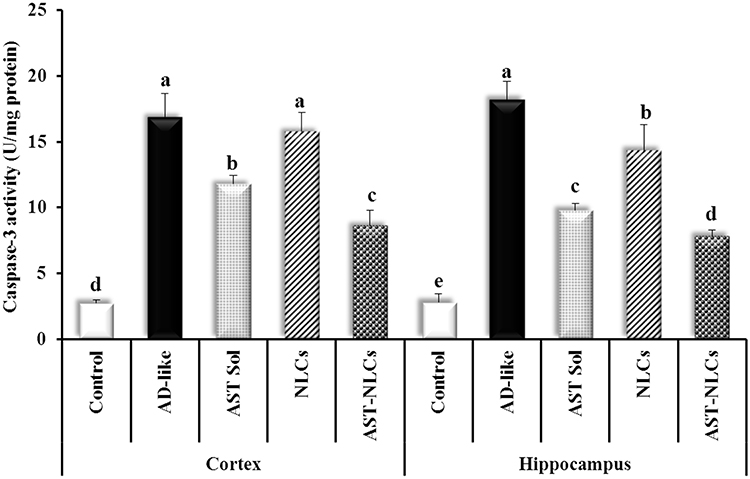 |
Figure 8 Caspase-3 activity in the cortex and hippocampus of the studied groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (cortex or hippocampus), means with any common letter are not significantly different, while means with totally different letters (from a-e) are significantly different at p < 0.05. Abbreviations: AD-like, Alzheimer’s disease-like; AST, astaxanthin; Sol, solution; NLCs, nanostructured lipid carriers. |
The apoptotic pathway is regulated by the anti-apoptotic cytokines like Bcl-2 (B-cell lymphoma-2), and the pro-apoptotic cytokines like Bax (BcL2-Associated X). Under apoptotic stimulation, the pro-apoptotic cytokines promote the release of cytochrome c (Cyt c) from the mitochondria into the cytoplasm. A complex comprising Cyt c, apoptotic protease activator-1 and caspase-9 then activates caspase-3, which triggers apoptosis. Bcl-2 inhibits the release of Cyt c and reduces apoptosis.97 In the present study, AlCl3-induced neurotoxicity is due to its ability to induce oxidative stress, ROS generation, mitochondrial dysfunction, and cytoskeleton changes that resulted in cell death necrosis or apoptosis, which is approved by significant activation of caspase-3 as a marker of apoptosis. Caspase-3 is an intracellular cysteine protease that exists as a pro-enzyme, becoming activated during the cascade of events associated with apoptosis.98,99 Treatment of AD-like rats with AST-NLCs was associated with anti-apoptotic activity as indicated by the decreased caspase-3 activity. Zhang et al100 showed that AST serves an important role in upregulation of Bcl-2 levels and downregulation of the activation of Cyt c, caspase-3, and Bax. Fan et al101 found that AST supplementation protects rats from homocysteine-related apoptosis through upregulation of Bcl-2 levels.
Some studies have shown that treatment using AST can promote nerve cell regeneration and increase gene expression of proteins important for brain recovery, such as glial fibrillary acidic protein (GFAP), microtubule associated protein 2 (MAP-2), brain-derived neurotrophic factor (BDNF) and growth-associated protein 43 (GAP-43). GFAP serves significant roles in the repair of CNS injury, promotion of cell communication and alleviation of BBB damage. MAP-2 can regulate microtubule growth and neuronal regeneration. BDNF is responsible for neuronal survival and growth and the differentiation of new neurons, whereas upregulation of GAP-43 stimulates the protein kinase pathway and promotes neurite formation, regeneration and plasticity.102 Wu et al103 showed that AST significantly enhanced the expression of BDNF in the brain of aging rats.
AD appears to be a consequence of several convergent factors including oxidative stress, inflammation, activation of amyloidogenic pathway, accumulation of toxic protein aggregates (Aβ 1–42), disturbed brain neurotransmitters, neurodegeneration. The commonly prescribed medications for AD, are only symptomatic that do not stop the progressive pathology of the disease.104 Given the fact of worldwide spread of AD, the search for protective and effective disease-modifying treatments against the development of AD is of great importance. From the results obtained in the present study, the neuroprotective mechanisms of action of AST could be conferred upon its anti-amyloidogenic, anti-cholinesterase, antioxidant, anti-neuroinflammatory and anti-apoptotic properties and it could be a promising candidate for prevention and management of neurological disorders like AD. AST has low oral bioavailability due to its high lipophilicity, low solubility in gastrointestinal tract fluids, and suffers from hepatic first pass metabolism leading to poor oral absorption.10 Therefore, the intranasal (IN) route was used in this study for direct brain targeting. IN administration avoids first pass metabolism and gastrointestinal inactivation, achieves direct nose to brain drug delivery via olfactory and trigeminal nerve pathways bypassing BBB, and indirectly into the systemic circulation before crossing the BBB to reach the brain, increases distribution and bioavailability of drugs in the brain, and therefore it could reduce the drug dose to be administered.23 AST has the ability to bypass the BBB. In this study, astaxanthin was loaded in lipid nanoparticles, NLCs, to improve the intranasal permeability and enhance the brain targeting.
It was observed from the results of pharmacodynamics study that AD-like rats treated intranasally with AST in the optimized NLC formulation showed significantly better in vivo effects compared with free drug solution (IN). This could be due to rapid clearance of the administered AST-solution from the nasal cavity by the mucociliary clearance mechanism, short contact time with nasal mucosa and enzymatic degradation of free drug in nasal cavity. NLCs offered significant enhancement in nose-to-brain delivery of AST, as they can reduce mucociliary clearance, increase the nasal mucosal contact due to the occlusive nature of lipids in NLCs, the incorporation of the drug in NLCs reduces enzymatic drug degradation within the nasal cavity better than free drug solutions, and they improve the drug’s nasomucosal permeability greater than free drug solutions due to their flexible nano-sized (mean PS = 142.8 nm, less than 200 nm) lipophilic particles that can easily squeeze through intercellular spaces between olfactory cells and transported directly to the brain, increase drug bioavailability in the brain.33,66 Several studies reported the direct nose to brain transport and the significant enhancement of drug delivery to the brain following IN administration of drug loaded NLCs.28,105,106
Astaxanthin was encapsulated in NLCs in two recent studies. Gautam et al39 developed AST-NLCs and incorporated in an in situ thermoreversible nasal gel for better neuronal uptake and efficacy for management of Parkinson’s disease. The average PS, ZP, and EE of the optimized AST-NLC formulation were 225.6 ± 3.04 nm, −52.64 mV, and 65.91 ± 1.22%, respectively. Pharmacokinetic studies showed better availability of AST in the brain on the rats treated with IN AST-NLC in-situ gel as compared to those treated with IN free AST-in-situ gel. AST-NLC gel (IN) had higher Cmax,brain (9.5-fold) and AUCbrain (7.79-fold) values than the free drug gel (IN). In haloperidol-treated rats, the AST-NLC gel (IN) improved rat behavior in the rotarod test and akinesia measurements as compared with the free drug gel (IN). Geng et al107 formulated AST-NLCs to improve its stability, water solubility, skin permeability and retention and reduce drug-related side effects.56 The optimized AST-NLC formulation had a mean PS of 67.4 ± 2.1 nm and 94.3 ± 0.5% EE. AST-NLCs were found to be nonirritating, homogenous, and with excellent stability and water solubility, and higher cumulative skin permeability and retention. This study concluded that NLCs serve as a promising carrier for site-specific targeting of AST with better stability and skin penetration.
Histopathological Analysis: Hematoxylin and Eosin (H and E) Staining
As shown in Figure 9, the histological analysis of the brain tissues of control rats revealed nearly normal cortical nerve cells. The cortical tissue of the AD-like untreated rats showed degenerated shrunken darkly stained neurons with increased pericellular spaces, necrotic neurons associated with satellitosis and neuronophagia, and increased glial cells. The histoarchitecture of the cortical tissues of the AD-like rats was markedly improved after IN treatment with AST in NLC formulation, associated with few numbers of degenerated and necrotic neurons.
 |
Figure 9 Histology of cerebral cortex stained with hematoxylin and eosin (X400) of (a) control rats, (b) Alzheimer’s disease (AD)-like untreated rats, (c) astaxanthin-solution (AST-Sol) treated rats, and (d) astaxanthin-nanostructured lipid carriers (AST–NLCs) treated rats. Normal neurons (black arrow), degenerated neurons (Orange arrow), necrotic neurons associated with satellitosis and neuronophagia (red arrows), and glial cells (yellow arrow). |
In Figure 10, the control rats showed normal hippocampal nerve cells. The hippocampal tissue of AD-like untreated rats showed distorted cellular morphology with increased pericellular space, misaligned degenerated neurons, pyknotic neurons, necrotic neurons associated with satellitosis, and neuronophagia and glial cells. The hippocampal nerve cell morphology showed marked improvement with AST-NLCs which was associated with a few numbers of degenerated and necrotic neurons.
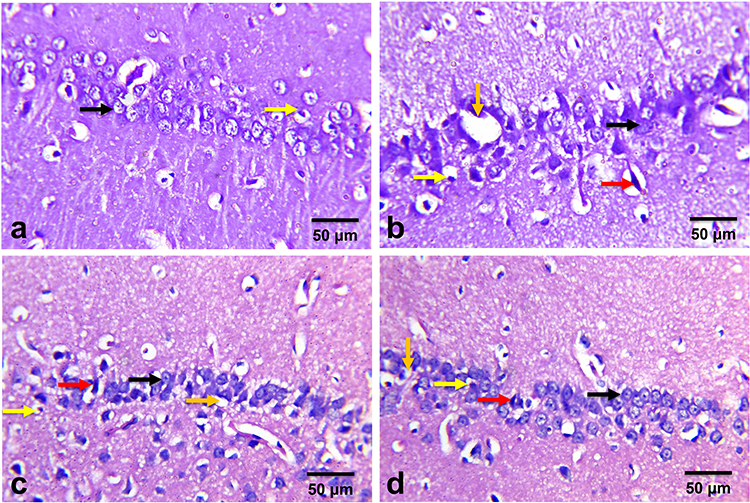 |
Figure 10 Histology of hippocampus stained with hematoxylin and eosin (X400) of (a) control rats, (b) Alzheimer’s disease (AD)-like untreated rats, (c) astaxanthin-solution (AST-Sol) treated rats, and (d) astaxanthin-nanostructured lipid carriers (AST–NLCs) treated rats. Normal neurons (black arrow), degenerated neurons (red arrow), glial cells (yellow arrow), and pericellular space (Orange arrow). |
Various histopathological alterations (neuronal degeneration and necrosis and diffuse gliosis) were observed in the cerebral and hippocampal tissues of AD-like untreated group were reliable with AlCl3-induced neurotoxicity and memory dysfunction which may be attributed to the ability of AlCl3 to induce oxidative stress, ROS generation, mitochondrial dysfunction, disruption of vital cellular processes and cytoskeleton changes which resulted in cell death necrosis.108 The cerebral and hippocampal lesions are consistent with those reported by Sumathi et al.109 The observed ameliorative effects of AST on histopathological lesions were associated with similar effects on the behavioral test and biochemical results. AD-like rats treated with AST-NLCs showed significant improvement in cognitive performance in MWM test and neuroprotective role as indicated in biochemical parameters in the brain of treated rats.
Treatment with AST-NLCs had anti-amyloidogenic effects that caused a significant decline in Aβ1-42 content in the cortex and hippocampus. The anti-amyloidogenic effect of AST may be mediated via a significant reduction in β-secretase (BACE-1) expression as the present data indicated or may be due to its potent antioxidant effects mediated by stimulation of endogenous antioxidant system. The present study indicated that AST-NLC treatment significantly corrected the MDA and GSH in the rat brain. Wu et al103 showed that AST significantly ameliorated the histopathological changes in the hippocampus and enhanced the expression of brain derived neurotrophic factor (BDNF) in the brain of aging rats.
Correlation Study
The Pearson correlation study (Table 7) indicated the following relationships: the cortical and hippocampal Aβ1-42 contents were positively correlated with AChE, NF-kB, BACE-1 expression, MDA, and caspase-3 activity and negatively correlated with ACh and GSH. The ACh contents were positively correlated with GSH and negatively correlated with AChE, NF-kB, MDA, caspase-3, and BACE-1 expression. The GSH content showed similar correlation pattern to ACh, while AChE, NF-kB, caspase-3, MDA, and BACE-1 expression showed opposite correlation patterns.
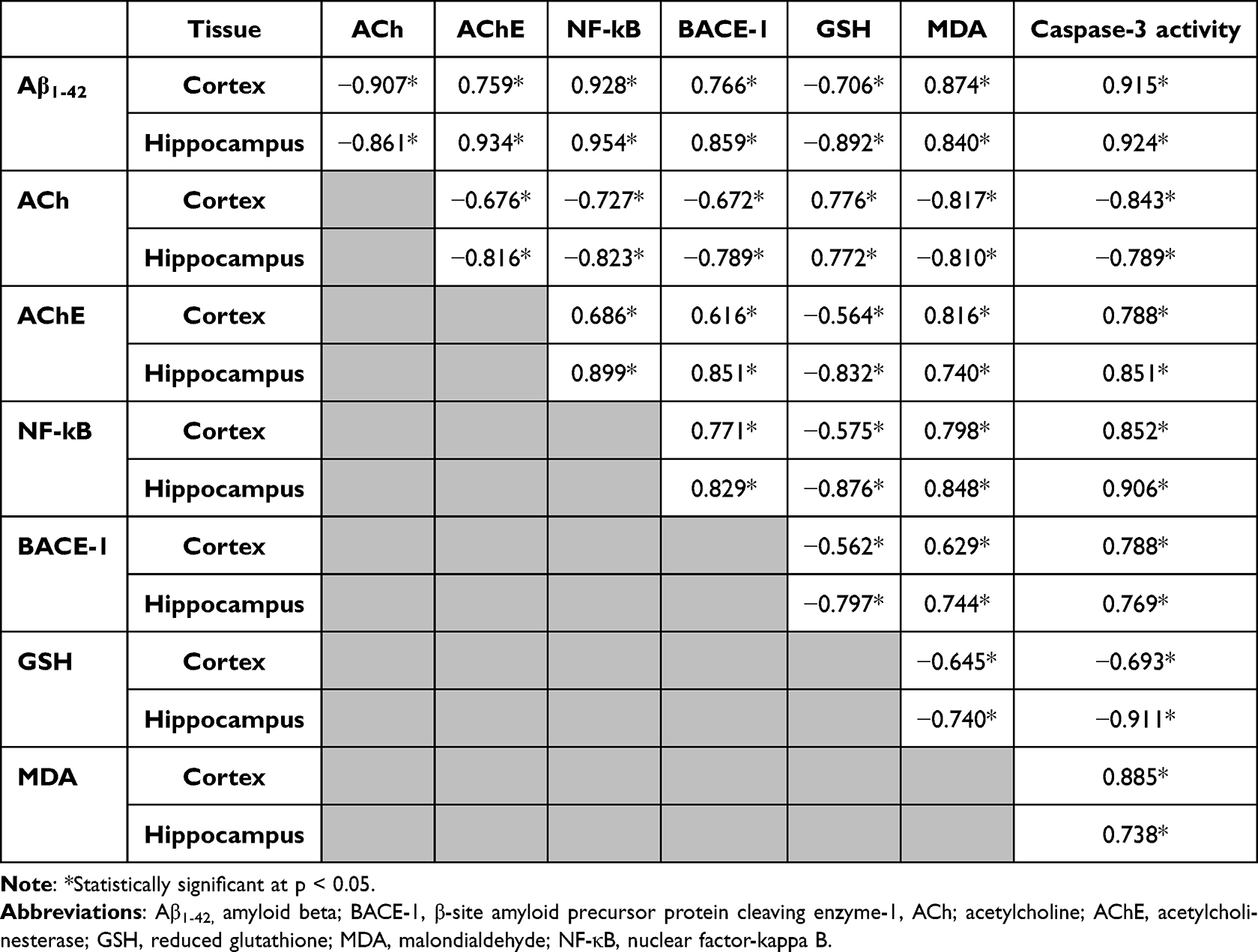 |
Table 7 Pearson Correlation (r) Study Between Different Parameters in AD-Like, AST-Sol, NLCs and AST-NLCs Groups (n = 24) in the Cortex and Hippocampus |
The correlation data confirmed the possible link between the amyloidogenic pathway progression and the disturbances in the cholinergic pathway; as the markers of amyloidogenic pathway (BACE-1 and Aβ1-42) were negatively correlated with ACh level and positively correlated with AChE activity, and also between the induced oxidative stress and induction of neuroinflammation as indicated by the positive correlation between MDA and NF-kB levels. Based on the above results and discussion, it is strongly suggested that AST could be a promising candidate for management of AD.
Conclusion
In this study, an optimized AST–NLC formulation was successfully prepared and showed a small mean particle size, high entrapment efficiency with biphasic release pattern; an initial rapid release followed by slow and sustained release for 24 h, and were stable when stored at 4–8 ±2°C. DSC indicated that AST existed in an amorphous state in the NLC matrix. Nose-to-brain delivery of AST-NLCs in AD-like rats showed anti-amyloidogenic, anti-cholinesterase, antioxidant, anti-neuroinflammatory and anti-apoptotic effects through targeting multiple pathways involved in AD progression including induced amyloidogenic pathway, impaired cholinergic neurotransmission, oxidative stress, neuroinflammation, and apoptosis, and correcting the cortical and hippocampal histopathology. Our results suggest that intranasal delivery of AST in NLCs could be a promising treatment for AD and circumvent its poor oral bioavailability.
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Carreiras MC, Mendes E, Perry MJ, Francisco AP, Marco-Contelles J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr Top Med Chem. 2013;13(15):1745–1770. doi:10.2174/15680266113139990135
2. Jahn H. Memory loss in Alzheimer’s disease. Dialogues Clin Neurosci. 2013;15(4):445–454. doi:10.31887/DCNS.2013.15.4/hjahn
3. Kales HC, Lyketsos CG, Miller EM, Ballard C. Management of behavioral and psychological symptoms in people with Alzheimer’s disease: an international Delphi consensus. Int Psychogeriatr. 2019;31(1):83–90. doi:10.1017/S1041610218000534
4. Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110(4):1129–1134. doi:10.1111/j.1471-4159.2009.06181.x
5. Kamat PK, Kalani A, Rai S, et al. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: understanding the therapeutics strategies. Mol Neurobiol. 2016;53(1):648–661. doi:10.1007/s12035-014-9053-6
6. Grimmig B, Kim SH, Nash K, Bickford PC, Douglas Shytle R. Neuroprotective mechanisms of astaxanthin: a potential therapeutic role in preserving cognitive function in age and neurodegeneration. Geroscience. 2017;39(1):19–32. doi:10.1007/s11357-017-9958-x
7. Ni Y, Nagashimada M, Zhuge F, et al. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: a comparison with vitamin E. Sci Rep. 2015;5:17192. doi:10.1038/srep17192
8. Higuera-Ciapara I, Félix-Valenzuela L, Goycoolea FM. Astaxanthin: a review of its chemistry and applications. Crit Rev Food Sci Nutr. 2006;46(2):185–196. doi:10.1080/10408690590957188
9. Zuluaga M, Gueguen V, Letourneur D, Pavon-Djavid G. Astaxanthin-antioxidant impact on excessive Reactive Oxygen Species generation induced by ischemia and reperfusion injury. Chem Biol Interact. 2018;279:145–158. doi:10.1016/j.cbi.2017.11.012
10. Taksima T, Chonpathompikunlert P, Sroyraya M, Hutamekalin P, Limpawattana M, Klaypradit W. Effects of astaxanthin from shrimp shell on oxidative stress and behavior in animal model of Alzheimer’s disease. Mar Drugs. 2019;17(11):628. doi:10.3390/md17110628
11. Ambati RR, Phang SM, Ravi S, Aswathanarayana RG. Astaxanthin: sources, extraction, stability, biological activities and its commercial applications–a review. Mar Drugs. 2014;12(1):128–152. doi:10.3390/md12010128
12. Shah MM, Liang Y, Cheng JJ, Daroch M. Astaxanthin-producing green microalga haematococcus pluvialis: from single cell to high value commercial products. Front Plant Sci. 2016;7:531.
13. Liu X, Luo Q, Rakariyatham K, et al. Antioxidation and anti-ageing activities of different stereoisomeric astaxanthin in vitro and in vivo. J Funct Food. 2016;25:50–61. doi:10.1016/j.jff.2016.05.009
14. EFSA Panel on Dietetic Products Nutrition Allergies. Scientific opinion on the safety of astaxanthin‐rich ingredients (AstaREAL A1010 and AstaREAL L10) as novel food ingredients. EFSA J. 2014;12(7):3757.
15. Choi HD, Kang HE, Yang SH, Lee MG, Shin WG. Pharmacokinetics and first-pass metabolism of astaxanthin in rats. Br J Nutr. 2011;105(2):220–227. doi:10.1017/S0007114510003454
16. Mercke Odeberg J, Lignell A, Pettersson A, Höglund P. Oral bioavailability of the antioxidant astaxanthin in humans is enhanced by incorporation of lipid based formulations. Eur J Pharm Sci. 2003;19(4):299–304. doi:10.1016/S0928-0987(03)00135-0
17. Østerlie M, Bjerkeng B, Liaaen-Jensen S. Plasma appearance and distribution of astaxanthin E/Z and R/S isomers in plasma lipoproteins of men after single dose administration of astaxanthin. J Nutr Biochem. 2000;11(10):482–490. doi:10.1016/S0955-2863(00)00104-2
18. Manabe Y, Komatsu T, Seki S, Sugawara T. Dietary astaxanthin can accumulate in the brain of rats. Biosci Biotechnol Biochem. 2018;82(8):1433–1436. doi:10.1080/09168451.2018.1459467
19. Chik MW, Mohd Affandi M, Singh GKS. Detection of astaxanthin at different regions of the brain in rats treated with astaxanthin nanoemulsion. J Pharm Bioallied Sci. 2022;14(1):25–30. doi:10.4103/jpbs.jpbs_464_21
20. Iwamoto T, Hosoda K, Hirano R, et al. Inhibition of low-density lipoprotein oxidation by astaxanthin. J Atheroscler Thromb. 2000;7(4):216–222. doi:10.5551/jat1994.7.216
21. Hanson LR, Frey WH. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9 Suppl 3(Suppl3):S5. doi:10.1186/1471-2202-9-S3-S5
22. Erdő F, Bors LA, Farkas D, Bajza Á, Gizurarson S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res Bull. 2018;143:155–170. doi:10.1016/j.brainresbull.2018.10.009
23. Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. doi:10.1002/jps.21924
24. Bahadur S, Pardhi DM, Rautio J, Rosenholm JM, Pathak K. Intranasal nanoemulsions for direct nose-to-brain delivery of actives for CNS disorders. Pharmaceutics. 2020;12(12):1230. doi:10.3390/pharmaceutics12121230
25. Musumeci T, Bonaccorso A, Puglisi G. Epilepsy disease and nose-to-brain delivery of polymeric nanoparticles: an overview. Pharmaceutics. 2019;11(3):118. doi:10.3390/pharmaceutics11030118
26. TdB C, Rios AC, Alves TFR, et al. Cachexia: pathophysiology and ghrelin liposomes for nose-to-brain delivery. Int J Mol Sci. 2020;21(17):5974. doi:10.3390/ijms21175974
27. Uppuluri CT, Ravi PR, Dalvi AV. Design, optimization and pharmacokinetic evaluation of Piribedil loaded solid lipid nanoparticles dispersed in nasal in situ gelling system for effective management of Parkinson’s disease. Int J Pharm. 2021;606:120881. doi:10.1016/j.ijpharm.2021.120881
28. Sarma A, Das MK, Chakraborty T, Das S. Nanostructured lipid carriers (NLCs)-based intranasal Drug Delivery System of Tenofovir disoproxil fumerate (TDF) for brain targeting. Res J Pharm Technol. 2020;13:5411–5424.
29. Lee D, Minko T. Nanotherapeutics for nose-to-brain drug delivery: an approach to bypass the blood brain barrier. Pharmaceutics. 2021;13(12):2049. doi:10.3390/pharmaceutics13122049
30. Akel H, Ismail R, Csóka I. Progress and perspectives of brain-targeting lipid-based nanosystems via the nasal route in Alzheimer’s disease. Eur J Pharm Biopharm. 2020;148:38–53. doi:10.1016/j.ejpb.2019.12.014
31. Haider M, Abdin SM, Kamal L, Orive G. Nanostructured lipid carriers for delivery of chemotherapeutics: a review. Pharmaceutics. 2020;12(3):288. doi:10.3390/pharmaceutics12030288
32. Fang CL, Al-Suwayeh SA, Fang JY. Nanostructured lipid carriers (NLCs) for drug delivery and targeting. Recent Pat Nanotechnol. 2013;7(1):41–55. doi:10.2174/187221013804484827
33. Alam MI, Baboota S, Ahuja A, Ali M, Ali J, Sahni JK. Intranasal administration of nanostructured lipid carriers containing CNS acting drug: pharmacodynamic studies and estimation in blood and brain. J Psychiatr Res. 2012;46(9):1133–1138. doi:10.1016/j.jpsychires.2012.05.014
34. Jazuli I, Annu NB, Nabi B, et al. Optimization of nanostructured lipid carriers of lurasidone hydrochloride using box-behnken design for brain targeting: in vitro and in vivo studies. J Pharm Sci. 2019;108(9):3082–3090. doi:10.1016/j.xphs.2019.05.001
35. Salem LH, El-Feky GS, Fahmy RH, El Gazayerly ON, Abdelbary A. Coated lipidic nanoparticles as a new strategy for enhancing nose-to-brain delivery of a hydrophilic drug molecule. J Pharm Sci. 2020;109(7):2237–2251. doi:10.1016/j.xphs.2020.04.007
36. Du W, Li H, Tian B, et al. Development of nose-to-brain delivery of ketoconazole by nanostructured lipid carriers against cryptococcal meningoencephalitis in mice. Colloids Surf B Biointerfaces. 2019;183:110446. doi:10.1016/j.colsurfb.2019.110446
37. Rajput A, Bariya A, Allam A, Othman S, Butani SB. In situ nanostructured hydrogel of resveratrol for brain targeting: in vitro-in vivo characterization. Drug Deliv Transl Res. 2018;8(5):1460–1470. doi:10.1007/s13346-018-0540-6
38. Bhatt PC, Srivastava P, Pandey P, Khan W, Panda BP. Nose to brain delivery of astaxanthin-loaded solid lipid nanoparticles: fabrication, radio labeling, optimization and biological studies. RSC Adv. 2016;6(12):10001–10010. doi:10.1039/C5RA19113K
39. Gautam D, Singh S, Maurya P, Singh M, Kushwaha S, Saraf SA. Appraisal of nano-lipidic astaxanthin cum thermoreversible gel and its efficacy in haloperidol induced parkinsonism. Curr Drug Deliv. 2021;18(10):1550–1562. doi:10.2174/1567201818666210510173524
40. Uner M. Preparation, characterization and physico-chemical properties of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC): their benefits as colloidal drug carrier systems. Pharmazie. 2006;61(5):375–386.
41. How CW, Abdullah R, Abbasalipourkabir R. Physicochemical properties of nanostructured lipid carriers as colloidal carrier system stabilized with polysorbate 20 and polysorbate 80. Afr J Biotechnol. 2011;10(9):1684–1689.
42. Cunha S, Costa CP, Loureiro JA, et al. Double optimization of rivastigmine-loaded Nanostructured Lipid Carriers (NLC) for nose-to-brain delivery using the Quality by Design (QbD) approach: formulation variables and instrumental parameters. Pharmaceutics. 2020;12(7):599. doi:10.3390/pharmaceutics12070599
43. Jores K, Mehnert W, Drechsler M, Bunjes H, Johann C, Mäder K. Investigations on the structure of solid lipid nanoparticles (SLN) and oil-loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95(2):217–227. doi:10.1016/j.jconrel.2003.11.012
44. Shrivastava A, Gupta VB. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron Young Sci. 2011;2(1):21–25. doi:10.4103/2229-5186.79345
45. Sun W, Xing L, Leng K, et al. Determination of astaxanthin in Antarctic krill and its products by high performance liquid chromatography. J Food Saf Qual. 2017;8(4):1248–1253.
46. Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A. Design and characterization of astaxanthin-loaded nanostructured lipid carriers. Innov Food Sci Emerg Technol. 2014;26:366–374. doi:10.1016/j.ifset.2014.06.012
47. Bunjes H, Unruh T. Characterization of lipid nanoparticles by differential scanning calorimetry, X-ray and neutron scattering. Adv Drug Deliv Rev. 2007;59(6):379–402. doi:10.1016/j.addr.2007.04.013
48. Chadha R, Bhandari S. Drug-excipient compatibility screening–role of thermoanalytical and spectroscopic techniques. J Pharm Biomed Anal. 2014;87:82–97. doi:10.1016/j.jpba.2013.06.016
49. Yasir M, Sara UV. Solid lipid nanoparticles for nose to brain delivery of haloperidol: in vitro drug release and pharmacokinetics evaluation. Acta Pharm Sin B. 2014;4(6):454–463. doi:10.1016/j.apsb.2014.10.005
50. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
51. Makoni PA, Wa Kasongo K, Walker RB. Short term stability testing of Efavirenz-Loaded Solid Lipid Nanoparticle (SLN) and Nanostructured Lipid Carrier (NLC) dispersions. Pharmaceutics. 2019;11(8):397. doi:10.3390/pharmaceutics11080397
52. Ali AA, Ahmed HI, Abu-Elfotuh K. Modeling stages mimic Alzheimer’s disease induced by different doses of aluminum in rats: focus on progression of the disease in response to time. J Alzheimer’s Parkinsonism Dementia. 2016;1(1):2.
53. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. doi:10.1038/nprot.2006.116
54. Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106(1):207–212. doi:10.1016/0003-2697(80)90139-6
55. Draper HH, Hadley M. Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 1990;186:421–431.
56. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
57. Lilli R, Bancroft J, Gamble M. The Hematoxylin and Eosin, Histopathologic Technique and Practical Biochemistry. 8th ed. London: Elsevier; 2008.
58. Zhang X, Liu J, Qiao H, et al. Formulation optimization of dihydroartemisinin nanostructured lipid carrier using response surface methodology. Powder Technol. 2010;197(1–2):120–128. doi:10.1016/j.powtec.2009.09.004
59. Alam T, Pandit J, Vohora D, Aqil M, Ali A, Sultana Y. Optimization of nanostructured lipid carriers of lamotrigine for brain delivery: in vitro characterization and in vivo efficacy in epilepsy. Expert Opin Drug Deliv. 2015;12(2):181–194. doi:10.1517/17425247.2014.945416
60. Azhar Shekoufeh Bahari L, Hamishehkar H. The Impact of Variables on Particle Size of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers; A Comparative Literature Review. Adv Pharm Bull. 2016;6(2):143–151. doi:10.15171/apb.2016.021
61. Sangsen Y, Laochai P, Chotsathidchai P, Wiwattanapatapee R. Effect of solid lipid and liquid oil ratios on properties of nanostructured lipid carriers for oral curcumin delivery. Adv Mater Res. 2015;1060:62–65. doi:10.4028/www.scientific.net/AMR.1060.62
62. Das S, Ng WK, Tan RB. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci. 2012;47(1):139–151. doi:10.1016/j.ejps.2012.05.010
63. Kovacevic A, Savic S, Vuleta G, Müller RH, Keck CM. Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): effects on size, physical stability and particle matrix structure. Int J Pharm. 2011;406(1–2):163–172. doi:10.1016/j.ijpharm.2010.12.036
64. Liu C-H, Wu C-T. Optimization of nanostructured lipid carriers for lutein delivery. Coll Surf a Physicochem Eng Aspect. 2010;353(2–3):149–156. doi:10.1016/j.colsurfa.2009.11.006
65. Khan S, Shaharyar M, Fazil M, Baboota S, Ali J. Tacrolimus-loaded nanostructured lipid carriers for oral delivery – Optimization of production and characterization. Eur J Pharm Biopharm. 2016;108:277–288. doi:10.1016/j.ejpb.2016.07.017
66. Madane RG, Mahajan HS. Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: design, characterization, and in vivo study. Drug Deliv. 2016;23(4):1326–1334. doi:10.3109/10717544.2014.975382
67. Subramaniam B, Siddik ZH, Nagoor NH. Optimization of nanostructured lipid carriers: understanding the types, designs, and parameters in the process of formulations. J Nanopart Res. 2020;22(6):1–29. doi:10.1007/s11051-020-04848-0
68. Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliv Rev. 2011;63(6):456–469. doi:10.1016/j.addr.2011.02.001
69. Hamdani J, Moës AJ, Amighi K. Physical and thermal characterisation of Precirol and Compritol as lipophilic glycerides used for the preparation of controlled-release matrix pellets. Int J Pharm. 2003;260(1):47–57. doi:10.1016/S0378-5173(03)00229-1
70. Zambito Y, Pedreschi E, Di Colo G. Is dialysis a reliable method for studying drug release from nanoparticulate systems? A case study. Int J Pharm. 2012;434(1–2):28–34. doi:10.1016/j.ijpharm.2012.05.020
71. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5(1):37–42. doi:10.1016/0168-3659(87)90035-6
72. Rahman SO, Panda BP, Parvez S, et al. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed Pharmacother. 2019;110:47–58. doi:10.1016/j.biopha.2018.11.043
73. Rajmohan R, Reddy PH. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J Alzheimers Dis. 2017;57(4):975–999. doi:10.3233/JAD-160612
74. Coronel R, Bernabeu-Zornoza A, Palmer C, et al. Role of Amyloid Precursor Protein (APP) and its derivatives in the biology and cell fate specification of neural stem cells. Mol Neurobiol. 2018;55(9):7107–7117. doi:10.1007/s12035-018-0914-2
75. Fanaee-Danesh E, Gali CC, Tadic J, et al. Astaxanthin exerts protective effects similar to bexarotene in Alzheimer’s disease by modulating amyloid-beta and cholesterol homeostasis in blood-brain barrier endothelial cells. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2224–2245. doi:10.1016/j.bbadis.2019.04.019
76. Yuan XZ, Sun S, Tan CC, Yu JT, Tan L. The role of ADAM10 in Alzheimer’s disease. J Alzheimers Dis. 2017;58(2):303–322. doi:10.3233/JAD-170061
77. Hafez HA, Kamel MA, Osman MY, Osman HM, Elblehi SS, Mahmoud SA. Ameliorative effects of astaxanthin on brain tissues of Alzheimer’s disease-like model: cross talk between neuronal-specific microRNA-124 and related pathways. Mol Cell Biochem. 2021;476(5):2233–2249. doi:10.1007/s11010-021-04079-4
78. Yassin N, El-Shenawy S, Mahdy KA, et al. Effect of boswellia serrata on Alzheimer’s disease induced in rats. J Arab Soc Med Res. 2013;8:1–11.
79. Van Beek AH, Claassen JA. The cerebrovascular role of the cholinergic neural system in Alzheimer’s disease. Behav Brain Res. 2011;221(2):537–542. doi:10.1016/j.bbr.2009.12.047
80. Kar S, Slowikowski SP, Westaway D, Mount HT. Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci. 2004;29(6):427–441.
81. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–383. doi:10.1038/s41580-020-0230-3
82. Goto S, Kogure K, Abe K, et al. Efficient radical trapping at the surface and inside the phospholipid membrane is responsible for highly potent antiperoxidative activity of the carotenoid astaxanthin. Biochim Biophys Acta. 2001;1512(2):251–258. doi:10.1016/S0005-2736(01)00326-1
83. Yang Y, Seo JM, Nguyen A, et al. Astaxanthin-rich extract from the green alga Haematococcus pluvialis lowers plasma lipid concentrations and enhances antioxidant defense in apolipoprotein E knockout mice. J Nutr. 2011;141(9):1611–1617. doi:10.3945/jn.111.142109
84. Li Z, Dong X, Liu H, et al. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated Phase II enzymes through activation of PI3K/Akt. Mol Vis. 2013;19:1656–1666.
85. Tripathi DN, Jena GB. Astaxanthin intervention ameliorates cyclophosphamide-induced oxidative stress, DNA damage and early hepatocarcinogenesis in rat: role of Nrf2, p53, p38 and phase-II enzymes. Mutat Res. 2010;696(1):69–80. doi:10.1016/j.mrgentox.2009.12.014
86. Nakagawa K, Kiko T, Miyazawa T, et al. Antioxidant effect of astaxanthin on phospholipid peroxidation in human erythrocytes. Br J Nutr. 2011;105(11):1563–1571. doi:10.1017/S0007114510005398
87. Suzer T, Coskun E, Demir S, Tahta K. Lipid peroxidation and glutathione levels after cortical injection of ferric chloride in rats: effect of trimetazidine and deferoxamine. Res Exp Med. 2000;199(4):223–229.
88. Akkoyun HT, Bengu AS, Ulucan A, et al. Effect of astaxanthin on rat brains against oxidative stress induced by cadmium: biochemical, histopathological evaluation. J Instit Sci Technol. 2018;8(4):33–39. doi:10.21597/jist.412070
89. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
90. Suzuki Y, Ohgami K, Shiratori K, et al. Suppressive effects of astaxanthin against rat endotoxin-induced uveitis by inhibiting the NF-kappaB signaling pathway. Exp Eye Res. 2006;82(2):275–281. doi:10.1016/j.exer.2005.06.023
91. Fakhri S, Dargahi L, Abbaszadeh F, Jorjani M. Astaxanthin attenuates neuroinflammation contributed to the neuropathic pain and motor dysfunction following compression spinal cord injury. Brain Res Bull. 2018;143:217–224. doi:10.1016/j.brainresbull.2018.09.011
92. Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol. 2020;98(1):28–41. doi:10.1111/imcb.12301
93. Park JS, Chyun JH, Kim YK, Line LL, Chew BP. Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans. Nutr Metab. 2010;7:18. doi:10.1186/1743-7075-7-18
94. Kim RE, Shin CY, Han SH, Kwon KJ. Astaxanthin suppresses PM2.5-induced neuroinflammation by regulating akt phosphorylation in BV-2 microglial cells. Int J Mol Sci. 2020;21(19):7227.
95. Han JH, Lee YS, Im JH, et al. Astaxanthin ameliorates lipopolysaccharide-induced neuroinflammation, oxidative stress and memory dysfunction through inactivation of the signal transducer and activator of transcription 3 pathway. Mar Drugs. 2019;17(2):123. doi:10.3390/md17020123
96. Che H, Li Q, Zhang T, et al. Effects of astaxanthin and docosahexaenoic-acid-acylated astaxanthin on Alzheimer’s Disease in APP/PS1 double-transgenic mice. J Agric Food Chem. 2018;66(19):4948–4957. doi:10.1021/acs.jafc.8b00988
97. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. doi:10.1038/nrm3722
98. Haider S, Liaquat L, Ahmad S, et al. Naringenin protects AlCl3/D-galactose induced neurotoxicity in rat model of AD via attenuation of acetylcholinesterase levels and inhibition of oxidative stress. PLoS One. 2020;15(1):e0227631. doi:10.1371/journal.pone.0227631
99. Rinwa P, Kumar A, Garg S, Scavone C. Suppression of neuroinflammatory and apoptotic signaling cascade by curcumin alone and in combination with piperine in rat model of olfactory bulbectomy induced depression. PLoS One. 2013;8(4):e61052. doi:10.1371/journal.pone.0061052
100. Zhang XS, Zhang X, Wu Q, et al. Astaxanthin alleviates early brain injury following subarachnoid hemorrhage in rats: possible involvement of Akt/bad signaling. Mar Drugs. 2014;12(8):4291–4310. doi:10.3390/md12084291
101. Fan CD, Sun JY, Fu XT, et al. Astaxanthin attenuates homocysteine-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage. Front Physiol. 2017;8:1041. doi:10.3389/fphys.2017.01041
102. Damodara Gowda KM, Suchetha Kumari N, Ullal H. Role of astaxanthin in the modulation of brain-derived neurotrophic factor and spatial learning behavior in perinatally undernourished Wistar rats. Nutr Neurosci. 2020;23(6):422–431. doi:10.1080/1028415X.2018.1515301
103. Wu W, Wang X, Xiang Q, et al. Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct. 2014;5(1):158–166. doi:10.1039/C3FO60400D
104. Knight R, Khondoker M, Magill N, Stewart R, Landau S. A systematic review and meta-analysis of the effectiveness of acetylcholinesterase inhibitors and memantine in treating the cognitive symptoms of dementia. Dement Geriatr Cogn Disord. 2018;45(3–4):131–151. doi:10.1159/000486546
105. Kumar N, Gupta GD, Arora D. DoE directed optimization, development and characterization of resveratrol loaded Nlc system for the nose to brain delivery in the management of glioblastoma multiforme. Res Square. 2021;1:1–23.
106. Jojo GM, Kuppusamy G, De A, Karri V. Formulation and optimization of intranasal nanolipid carriers of pioglitazone for the repurposing in Alzheimer’s disease using Box-Behnken design. Drug Dev Ind Pharm. 2019;45(7):1061–1072. doi:10.1080/03639045.2019.1593439
107. Geng Q, Zhao Y, Wang L, Xu L, Chen X, Han J. Development and Evaluation of Astaxanthin as Nanostructure Lipid Carriers in Topical Delivery. AAPS PharmSciTech. 2020;21(8):318. doi:10.1208/s12249-020-01822-w
108. Buraimoh A, Bello A, Ojo SA, Hambolu JO, Adebisi SS. Effects of aluminium chloride exposure on the histology of the cerebral cortex of adult Wistar rats. J Biol Life Sci. 2012;3(1):87–113. doi:10.5296/jbls.v3i1.1421
109. Sumathi T, Shobana C, Thangarajeswari M, Usha R. Protective effect of L-Theanine against aluminium induced neurotoxicity in cerebral cortex, hippocampus and cerebellum of rat brain – histopathological, and biochemical approach. Drug Chem Toxicol. 2015;38(1):22–31. doi:10.3109/01480545.2014.900068