Comparative Analysis of the Interaction of Silver Nanoparticles with Hexokinase from Trypanosoma brucei and Humans

Introduction
Trypanosomiasis (African sleeping sickness), the fatal, yet treatable infectious disease, is caused by the parasite Trypanosoma brucei and, despite continual efforts by the World Health Organization (WHO) on prevention and treatment of the disease, reported cases continue to increase.1–3 Current treatment suffers from a general lack of anti-trypanosomal therapy, excessive toxicity, increased resistance by trypanosomal strains, and unsuccessful efforts at vaccine development.4 Consequently, there is a continued pressing requirement for the development of inventive strategies to combat this disease. If such drugs or, specifically, anti T. brucei protocols are to be realized then a thorough understanding of the parasites life cycle is paramount. It is not the intention in this paper to report on this life cycle in any detail as literature reports abound. Nevertheless, since the Blood Stream Form (BSF) of the parasite depends entirely on glycolysis for its production of energy, it is clear that any enzymes within this cycle are a potential target for such anti-trypanosomals.
Interference with any of these enzymes would starve the parasite of cellular energy.5–8 One such enzyme is hexokinase that is responsible for the rate-limiting phosphorylation of glucose into glucose-6-phosphate.
Nanotechnology is capable of exploiting pathophysiological circumstances and anatomical variations that already exist in unhealthy tissues and enables the development of therapeutic agents with altered therapeutic and pharmacological properties that overcome many of the limitations associated with conventional treatments9–11 including quantification of HIV-1.12–14
Nanoparticles, especially AgNPs, have been studied broadly for use in bionanomedical applications.15–18 Our own laboratories have revealed an enhanced yield in the synthesis of metal (gold, silver, platinum) nanoparticles and their role in an improved activity of ferroxidase and ATPase of GroEL chaperonin.19 Furthermore, we have presented their effect against Arginine kinase from Trypanosoma brucei,20 against Triose-phosphate-isomerase of P. falciparum,21 and their interaction with neuronal Nitric oxide synthase and the fibrillogenesis mechanism in Alzheimer’s disease.22 Other research has developed these metal nanoparticles as agents for treating carcinomas, lymphomas, control infections neurological disorders, and diabetes.23–25
A common mechanism that underpins these nanomedical and/or nanotechnological associations with enzymes has been the interactions of thiol groups of sulfur bearing amino acids (cysteine/methionine) with nanoparticles of silver and gold.26 Consequently the selective inhibition of similarly functioning enzymes, whether they are from mammalian or parasitic origins, may be exploited as a potential therapy in clinical trials. Synthesizing nanoparticles to selectively interact with enzymes is a new and exciting research direction in the application for biomedicine and follows two distinct paradigms: a) the creation of various non-specific binding of the nanoparticle to the thiol groups resulting in loss of enzyme activity, usually associated with conformational changes to the enzyme structure; and b) a non-covalent technique where “naked” functionalized nanoparticles that interact with enzymes under standard protein–protein interactions through the formation of an enzyme “corona” around the nanoparticles. Either mechanism leads to structural changes of the enzyme and as the particle gets smaller the protein compositions or its interactions change. The “nanoparticle-enzyme corona” has entirely different biological properties as compared to the native enzyme.
The present study, therefore, focuses on the expression, purification, and characterization of a hexokinase from TbHK and the human isoform HsGCK followed by a comparative interaction with silver nanoparticles. The differences in primary amino acid sequences between these two kinases can be exploited for selective targeting.
Materials and Methods
Materials
Genomic DNA of T. brucei, a gift from Professor Ullman (Oregon Health Sciences University, USA), was employed as a template for PCR amplification. All spectrophotometric analyses were performed in 96-well micro-titre plates (BioTek Synergy MX spectrophotometer using Gen5™ 2.0 Data Analysis Software); liquid chromatography was performed on an Äkta FPLC Purification System (GE Healthcare Life Sciences AB, Uppsala, Sweden) according to the manufacturer’s instructions; polymerase chain reactions (PCR) were carried out using a T100 thermal cycler (Biorad, South Africa); a Bioflux Biospin kit was obtained from Separations Scientific (South Africa). CloneJet® PCG cloning kit was obtained from ThermoFisher Scientific (South Africa) and pGEM-T® Easy vector system from Promega Corporation.
Cloning, Expression, and Purification of HsGCK and TbHK
The oligonucleotides used for amplification of the target TbHK were obtained from the gene sequence (NCBI: XM_817363.1) while those for HsGCK were from Addgene Inc (plasmid #23750).27 Primers (reverse and forward) were designed using the ORFs represented in Table 1.
 |
Table 1 Oligonucleotide Primers Used for PCR Amplification and Sequence PCRs for TbHK and HsGCK. Underlined Sequences Indicate the Introduced Restriction Endonuclease Sites for NdeI (Forward) and XhoI (Reverse) |
The PCR reaction for TbHK gene was carried out in 50 μL (total volume): dNTP (10 mM, 1 μL), DNA template (50 ng), Phusion hot start II DNA pol. (0.5 μL, 0.02 U), TbHK – NdeI (10 μM, 0.2 μL), TbHK – XhoI (10 μM, 0.2 μL), Phusion High Fidelity buffer (5x, 10 μL), and distilled water. PCR conditions were carried out according to Table 2. The PCR reaction for the HsHK gene was carried out in 50 μL (total volume): dNTP (10 mM, 1 μL), DNA template (pDORN223-GCK plasmid, 50 ng), Phusion hot start II DNA polymerase (2.6 U), HsGCK – NdeI (10 μM, 1 μL), HsGCK – XhoI (10 μM, 1 μL), Phusion High Fidelity buffer containing MgCl2 (10x, 5 μL), distilled water. A two-step PCR protocol was carried out according to Table 2.
 |
Table 2 The PCR Protocols for TbHK and HsGCK Gene Amplification |
The products were then purified by agarose gel electrophoresis (0.8%, 100 V, 60 min) in TAE buffer (0.04 M Tris-HCl, 1 mM EDTA pH 8.0, and 0.021 mM glacial acetic acid) containing 0.6 μg.mL−1 ethidium bromide. The products were visualized under low UV radiation (Darkreader, BioLabo, Switzerland) and the applicable bands excised from the gel and extracted using the BioFlux BioSpin gel extraction kit.
Purity, concentration, and integrity of the nucleic acids were determined at 260 nm, using a NanoDrop-2000 spectrophotometer (Thermal Scientific, USA).
Purified TbHK and HsGCK PCR products were then ligated into CloneJet® and pGEM-T® cloning vector systems. For TbHK, the ligation reaction (4°C, 16 h) was performed in a 20 µL reaction volume consisting of 2x ligation buffer (10 µL), CloneJet® vector (50 ng), PCR product (8 µL), and T4 DNA ligase (0.3 Weiss units). For HsGCK, the ligation reaction (4°C, 16 h) was performed in a 20 µL total reaction volume consisting of 10x ligation buffer (2 µL), pGEM-T® vector (50 ng), 6 µL PCR product, and T4 DNA ligase (0.3 Weiss units). The ligation mixtures were subjected to a double digest with the restriction enzymes (NdeI and XhoI) before being purified. E. coli TOP10 (Invitrogen), JM109 (Promega), and BL21(DE3) containing the pRARE2 (Lucigen) plasmid were made competent chemically using the protocol described in literature28 with slight modifications. A colony of E. coli cells was inoculated into LB medium (5.0 mL) and incubated with aeration (18 h, 37°C). This culture (1.0 mL) was then inoculated into SOB media (5 g.l−1, yeast extract; 20 g.l−1, tryptone; 10 mM NaCl, KCl, MgCl2, and MgSO4) and incubated at 37°C until OD600 reached 0.600. The cell culture was cooled (ice-water, 15 min), centrifuged (3,000 x g, 5 min, 4°C), and the pellet re-suspended in TfBI buffer (40 mL, pH 5.8; 30 mM CH3COOK, 100 mM RbCl, 10 mM CaCl2, 50 mM MnCl2, and 15% glycerol v/v). After a further 15 minutes the cells were centrifuged (3,000 x g, 5 min, 4°C) and the pellet re-suspended in TfBII buffer (4 mL, pH 6.5; 10 mM MOPS, 75 mM CaCl2, 10 mM RbCl2 and 15% glycerol v/v). After a final 15 minutes on ice the suspension was stored at –80°C in aliquots (50 μL).
The ligated TbHK-CloneJet® (0.5 μL) and HsGCK-pGEM-T® (2.5 μL) constructs were then inserted into pET28b(+) [for N-terminal His-tagged fused proteins] and pET22b(+) [for C-terminal His-tagged fused proteins] expression vectors (Thermal Scientific). A reaction mixture (10 µL) consisted of 10x ligation buffer (1 μL), expression vector (50 ng), and T4 DNA ligase (0.75 Weiss units). The ligation mixtures were then incubated at 4°C overnight. Each ligated mixture [TbHK-CloneJet®-pET28b(+), TbHK-CloneJet®-pET22b(+), and HsGCKpGEM-T®-pET22b(+)] was transformed to the competent E. coli JM109 cells (100 μL) and E. coli TOP10 cells (100 μL), cooled on ice (30 min), heated (42°C, 40 sec), and then finally cooled again (ice, 2 min). These transformation mixtures were incubated (37°C, 60 min) with SOB media (250 μL) (5 g.l−1, yeast extract; 20 g.l−1 tryptone, 10 mM NaCl, KCl, MgCl2, MgSO4, and 20 mM glucose). The TbHK-CloneJet®-pET28b(+)-JM109 and TbHK-CloneJet®pET22b(+)-JM109 (200 μL) were plated onto LB agar plates containing kanamycin (30 μg.mL−1)/chloramphenicol (34 μg.mL−1) and ampicillin/chloramphenicol (100 μg.mL−1), respectively, and incubated (37°C, 16 h) before positive transformants were inoculated into LB media (5.0 mL), containing respective antibiotics, and the cultures incubated further (37°C, 16 h). The HsGCKpGEM-T®-pET22b(+)-TOP10 cultures (200 μL) were plated onto LB agar plates containing 100 μg.mL−1 ampicillin, 120 μg.mL−1 isopropyl-β-D-thiogalactoside (IPTG), and 80 μg.mL−1 5-bromo-4-chloro-3-indolyl-β-D-galactoside and incubated (37°C, 16 h) before positive transformants were inoculated into LB media (5.0 mL), containing ampicillin (100 μg.mL−1) and the cultures incubated further (37°C, 16 h). The plasmid DNA were isolated using the Biospin Plasmid Extraction Kit (BioFlux, Bioer Technology Co., Ltd) according to the manufacturers’ instruction. The plasmids were double digested using NdeI and XhoI restriction enzymes.
Reaction mixtures (20 µL) containing NdeI (1 μL), XhoI (2 μL), 10x buffer O (2 µL), and plasmid (50 ng) were incubated (37°C, 16 h) and the products examined by agarose gel electrophoresis. Each ligated mixture [TbHK-CloneJet®-pET28b(+), TbHK-CloneJet®-pET22b(+), and HsGCKpGEM-T®-pET22b(+)] was transformed to competent E. coli BL21(DE3) cells (100 μL) containing the pRARE2 plasmid, cooled on ice (30 min), heated (42°C, 40 sec), and then finally cooled again (ice, 2 min). These transformation mixtures were incubated (37°C, 60 min) with SOB media (250 μL) (5 g.L−1, yeast extract; 20 g.L−1, tryptone; 10 mM NaCl, KCl, MgCl2, MgSO4, and 20 mM glucose). The TbHK-CloneJet®-pET28b(+)-BL21(DE3) and TbHKCloneJet®-pET22b(+)-BL21(DE3) (200 μL) were plated onto LB agar plates containing, kanamycin (30 μg.mL−1)/chloramphenicol (34 μg.mL−1) and incubated (37°C, 16 h) before positive transformants were inoculated into LB media (5.0 mL), containing antibiotics, and the cultures incubated further (37°C, 16 h). The HsGCK-pGEM-T®-pET22b(+)-TOP10 cultures (200 μL) were plated onto LB agar plates containing 100 μg.mL−1 ampicillin, 120 μg.mL−1 isopropyl-β-D-thiogalactoside (IPTG), and 80 μg.mL−1 5-bromo-4-chloro-3-indolyl-β-D-galactoside and incubated (37°C, 16 h) before positive transformants were inoculated into LB media (5.0 mL), containing ampicillin (100 μg.mL−1) and the cultures incubated further (37°C, 16 h). The DNA in the plasmids were isolated using the Biospin Plasmid Extraction Kit (BioFlux, Bioer Technology Co., Ltd) according to the manufacturer’s instruction.
Each of the TbHK-CloneJet®-pET28b(+)-BL21(DE3), TbHK-CloneJet®-pET22b(+)-BL21(DE3), and HsGCK-pGEM-T®-pET22b(+)-BL21(DE3) constructs (1.0 mL) were inoculated into LB media (50 mL) containing, respectively, kanamycin (30 μg.mL−1), chloramphenicol (34 μg.mL−1), and ampicillin/chloramphenicol (100 μg.mL−1) and incubated (37°C, 16 h). These respective pre-cultures were then inoculated into 1 L of ZYP/5052 auto-induction media composed of 928 mL of ZY media (10 mg.L−1 tryptone, 5 mg.L−1 yeast extract), 50 mL of 20× NPS [0.5 M (NH4)2SO4, 1 M KH2PO4, 1 M Na2 HPO4], 20 mL of 50×5,052 (250 g.L−1 glycerol, 25 g.L−1 glucose, 100 g.L−1 α-lactose), 2 mL of 1 M MgSO4 supplemented, respectively, with kanamycin (30 μg.mL−1)/chloramphenicol (34 μg.mL−1), and ampicillin/chloramphenicol (100 μg.mL−1) and incubated (20°C, 36 h). Levels of protein expression were determined by SDS-PAG electrophoresis.
The cells were harvested by centrifugation (6,000 × g, 4°C, 15 min), washed 3x (Tris.HCl, 50 mM, pH 8.0), re-suspended in buffer containing lysozyme (1 mg.mL−1) and protease-inhibitor cocktail, then disrupted by freeze-thaw (−80°C/4°C; 2 cycles). Cell-debris were removed by centrifugation (3,000 x g, 4°C, 30 min), and the supernatant fractionated by ultra-centrifugation (100,000 x g, 4°C, 90 min). The final obtained supernatant was loaded onto a Ni-affinity column equilibrated with Tris.HCl (20 mM, pH 7.4) containing imidazole (20 mM) and NaCl (500 mM) and the fused proteins eluted with increasing concentrations of imidazole (0–500 mM). Selected fractions were collected, concentrated (Vivaspin, GE Healthcare, Sweden), and then the concentrate was purified by FPLC on Sephacryl S100HR column (1.6 x 100 cm) with Tris.HCl (50 mM, pH 7.4) containing NaCl (50 mM) and glycerol (15%) at a flow rate of 1 mL.min−1. Selected fractions were pooled, dialysed, concentrated, and then subjected to SDS PAGE and hexokinase activity.
HK Assay
HK activity was determined in a 96-well microtitre plate. Tris-HCl buffer (50 mM, pH 8.0, 210 μL) containing MgCl2 (13.3 mM), ATP (10 μL, 16.5 mM), NAD+ (10 μL, 6.8 mM), and glucose (670 μM, 50 µL) was incubated (30°C, 5 min) with enzyme extract (10 μL). Glucose-6-phosphate dehydrogenase (G6PDH) (10 μL, 300 U.mL−1) was then added and the absorbance increase at 340 nm monitored until no further change was observed. One unit of activity reduces 1 µmol glucose per minute at 30°C and pH 8.0.
Protein Concentration
The protein concentration for all experiments was routinely determined, using the bicinchoninic (BCA) assay (Pierce, Thermo Scientific); the assays were conducted in triplicate. Enzyme extract (50 μL) was added in a 1.5 mL Eppendorf tube followed by 1 mL of BCA reagent. The mixture was incubated at 37°C for 30 minutes. After the incubation, the absorbance of the solution was measured at 562 nm. The concentration of the unknown samples were determined using a BSA standard curve.
Silver Nanoparticles
Synthesis
The AgNPs were synthesised by established protocols.29 Ethanolic polyvinylpyrrolidone (1% (w/v), 10 mL) and silver nitrate (0.2 mL, 100 mM) were microwaved (450 MHz, 700 W, 5 sec) and the resulting pale yellow solution filtered through a 0.22 µM filter.
Characterization
UV/Vis spectroscopy was utilized, the absorption spectra (300–800 nm) of the nanoparticles were then obtained to assess the Plasmon bands associated with the AgNPs.
Transmission electron microscopy (TEM). AgNPs samples were prepared by placing a drop of the nanoparticle sample onto carbon-coated copper grids. Excess sample were removed after a few minutes using blotting paper. Before analysis the grids air-dried. The mean particle size and standard deviations (SD) were determined by analysing 200 particles using the computer software Java-based image processing program ImageJ.
Kinetic Analysis of Interaction of Ag Nanoparticles with HK
Enzyme solutions TbHK or HsGCK (10 µL) in 50 mM Tris-HCl buffer (pH 8.0) containing 13.3 mM MgCl2, 10 μL of 16.5 mM ATP (16.5 mM), and 10 μL of 6.8 mM NAD+ were incubated with 0–300 μM final concentration glucose at 30°C, in the absence and presence of silver nanoparticles (100 nM; final concentration) in a total volume of 300 µL. The HK activity was monitored by an increase in absorbance at 340 nm at each of the substrate concentrations.
Statistical Analyses
All analyses were done in triplicates and values reported as the means with standard deviation, p < 0.05 versus controls. Where it was necessary, analysis of variance was conducted using Statistica for Windows, version 8 (Statsoft Inc.), and Microsoft Excel 2010.
Results and Discussion
Expression and Purification of TbHK and HsGCK
The TbHK and HsGCK genes were both PCR amplified and showed, after agarose electrophoresis, a single band of 1,400 bp (Figure 1A and B) and the products ligated into CloneJet® and pGEM-T®-Easy cloning vectors, respectively (Figure 2A and B), followed by insertion, also respectively, into pET28b(+) [for N-terminal His-tagged fused proteins] and pET22b(+) [for C-terminal His-tagged fused proteins] expression vector systems (Figure 3A and B). Each ligated mixture [TbHK-CloneJet®-pET28b(+), and HsGCK-pGEM-T®-pET22b(+)] was transformed, respectively, to the competent E. coli JM109 cells and E. coli TOP10 cells.
 |
Figure 1 Agarose gel electrophoresis of the amplified gene: (A) TbHK; (B) HsGCK. |
 |
Figure 2 (A) CloneJet® plasmid containing TbHK gene digested with NdeI and XhoI. Lane 1 represents MassRuler™ DNA LadDer Standards (Thermo Scientific) and lanes 4 to 8 represent cloned plasmids screened for gene inserts. (B) pGEM-T® easy plasmid containing hGCK gene digested with NdeI and XhoI. Lane 1 represents the marker and lanes 2 to 6 represent cloned plasmids screened for gene inserts. |
 |
Figure 3 (A) pET28b(+) plasmid containing TbHK gene, double digested with XhoI and NdeI restriction enzymes. Lane 1 represents the DNA ladder, lanes 3 to 6 represents positive clones. (B) pET22b(+) plasmid containing HsGCK gene, double digested with XhoI and NdeI restriction enzymes. Lane 1 represents the DNA ladder, lanes 2 to 7 represent positive clones. |
The expressed enzymes were collected and purified initially through Nickel affinity column, followed by a Sephacryl S100HR gel filtration column. The elution profiles (Figure 4A) indicated, for TbHK, a large peak eluting at 80ml which had no hexokinase activity, and a smaller peak around 90ml reflecting positive for enzyme activity on a Zymogram (Figure 4B). With HsGCK, a major peak, eluting after 90-110mls (Figure 4C) was identified with glucokinase activity on a Zymogram (Figure 4C). All fractions containing hexokinase and glucokinase activity were confirmed by SDS-PAGE (Figure 5A and B).
 |
Figure 4 Elution profiles obtained from Sephacryl S-100HR resin and Zymogram analysis when loaded with (A and B) TbHK, (C and D) HsGCK. The area between the two arrows represents the region that was pooled for downstream purification. |
 |
Figure 5 SDS-PAGE analysis of (A) TbHK and (B) hGCK purification strategies. On both images (M) Marker, (2) lysate, (3) soluble fraction, (4) IMAC, (5) SEC, (6) Dialysis; showing the 25 kDa and 55 kDa purified TbHK and hGCK proteins, respectively. |
Analysis of the purification profiles for TbHK and HsGCK indicates that, after purification, the total protein decreased from 291 mg to 7.3 mg (for TbHK) and 1,275 mg to 45 mg (for HsGCK), as did the enzyme activities from 1.47 U to 0.21 U (for TbHK) and 40 U to 1.44 U (HsGCK). After a final dialysis the enzymes were produced in 12.5% yield and 31.6 fold purity (TbHK) and 5.6% yield and 20.4 fold purity (HsGCK), as presented in Table 3 and Table 4
 |
Table 3 Purification for TbHK |
 |
Table 4 Purification for HsGCK |
Kinetic Studies
Both TbHK and HsGCK followed Michaelis-Menten kinetics (Figure 6A). The parameters (Km, Vmax, kcat) and catalytic efficiency were estimated from equations (1), (2) and (3) as well as by non-linear regression and Hanes-Woolf plot (Figure 6B) at variable substrate (0–300 μM). There was a typical increase in enzyme activity with an increase in substrate concentration with Vmax and Km estimated, respectively, at 72 nmol.mL−1.min−1 and 46.7 µM for TbHK and 23 nmol.mL−1.min−1 and 4.5 μM for HsGCK. The turnover number and catalytic efficiency were, respectively, 2.2 min−1 and 0.047 µmol−1.min−1 for TbHK and 7.85 min−1 and 1.74 µmol−1.min−1 for HsGCK. The concentration of each enzyme (Et) was 33.0 μM (TbHK) and 2.93 μM (HsGCK)
(1)
(2)
(3)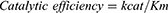
 |
Figure 6 (A) Michaelis Menten; (B) Hanes Woolf plots showing the kinetic analyses for TbHK and HsGCK in the absence and presence of silver nanoparticles. Data was obtained from 3 data sets, each collected on different days and in triplicate. Error bars represent standard deviation among data sets. |
The superior efficiency of HsGCK over TbHK is confirmed by the greater turnover rate and catalytic efficiency values.
Interaction of AgNPs with TbHK and HsGCK
The synthesized silver nanoparticles were characterized and visualized by transmission electron microscopy and UV/Vis spectroscopy in terms of frequency, size distribution (Figure 7), and surface plasmon resonance band. Their properties were in agreement with those reported elsewhere.30 The particles were spherical, with the majority monodispersed at 5–8 nm diameter. Literature suggests nanoparticles that are smaller than 20 nm are more effective for interactions with proteins.31 The affinity of the enzymes for AgNPs was determined over a substrate range (0–300 μM) by including the particles to 100 nM final concentration. The results indicated that, with TbHK, the Km increased from 39.3 to 54 µM, Vmax and kcat / Km decreased from 0.066 to 0.033 µmol.min−1.mL−1 and 0.0526 to 0.0380 min−1.µmol−1, representing a 38% reduction in the enzyme affinity with its substrate, a 50% reduction and 28% reduction in its activity and efficiency, respectively. These results illustrate a non-competitive type of inhibition mechanism by the AgNP’s. For HsGCK there was an observed significant decrease in the Km value from 4.5 mM to 3 mM, representing a 33% increase in the affinity. There was a prominent slight decrease in the Vmax value from 0.032 μmol.mL−1 to 0.029 μmol.min−1.mL−1, representing a 9% decrease in the maximal enzyme activity (Vmax), and a notable increase in the enzyme efficiency (kcat / Km) from 2.5 min−1 to 3.75 min−1 representing a 50% increase. The observed pattern of HsGCK and AgNPs falls under the uncompetitive inhibition.
 |
Figure 7 (A) Microwave assisted silver NPs synthesis, showing solution immediately after microwaving and 24 hours after microwaving. (B) Visible absorbance spectrum of silver nanoparticles indicating the surface plasmon resonance band at 421 nm. (C) TEM image of silver nanoparticles [Scale bar = 200 nm]; (D) frequency distribution of size of silver nanoparticles. |
Discussion
It is apparent from the experimental data that 0.1 μM silver NPs selectively inhibit TbHK hexokinase over hGCK. Remindful of the propensity of silver for sulfur and from our work on other biomedical targets with metal nanoparticles, it is expected that the AgNP associated with the hexokinase and/or glucokinase enzymes through sulfur bearing moieties such as cysteine and methionine residues.32 Consequently, sulfur-containing groups present on the surface of an enzyme would be anticipated to interact with AgNPs more favorably than if these groups were found buried within the enzyme. The observed selective inhibition of TbHK by AgNPs offers an exciting possibility for its utilization in the development of new anti-trypanosomal agents. This could be applied two-fold; firstly, the silver nanoparticles may be conjugated to an already existing anti-trypanosomal chemotherapeutic compound. This approach may improve a targeted delivery of the agent to hardly accessible areas, such as the brain. This may ease an existing major challenge in the treatment of trypanosomiasis. Secondly, the unconjugated NPs may be used to selectively inhibit the parasite hexokinase, which is critical for parasite survival.
In enhancing the access of drugs to the central nervous system (CNS), several drug carriers and hyperosmolar substances, including liposomes,33 lithium chloride, and sucrose,34 have been studied. The accumulation of poly(methylmethacrylate) (PMMA) NPs in rat brain demonstrating their capacity to cross the blood–brain barrier have been reported.35 Elsewhere, anti-cancer drugs dalagin and loperamide have been successfully delivered to the brain after their functionalization to NPs.36
The second stage of HAT presents with a similar challenge of the difficulty to get the anti-trypanosomal agents across the blood–brain barrier. It is reported that diminazine diaceturate, an anti-trypanosomal agent, was delivered to the CNS of mice by formulating a lipid-drug conjugate (LDC)-nanoparticle complex.37 Daunorubicin conjugated to polycyanocrylate NPs have enhanced entry into trypanosoma parasite, premaquine, and pentamidine combined with NPs have proved beneficial in the experimental visceral leishmaniasis.38 Knowledge from literature and data from this study suggest the inhibition of TbHK by unconjugated AgNPs may enhance the effect of the anti-trypanosomal agent. The anti-trypanosomal agent could further be functionalized to the NP, this may double the efficiency in killing the parasite with possibly few to no side-effects to the human hosts.
Results from this study may also suggest a possible treatment for the reduction of hyperglycemia observed in maturity onset diabetes of the youth (MODY)39 as the hGCK efficiency was enhanced. Glucokinase has been confirmed to be an important target in the treatment of diabetes, as it possesses a high association to the control of glucose levels.40,41 Studies have been conducted where solid lipid NPs, [poly(ε-caprolactone)],42 and dextran NPs have been pre-loaded with insulin and utilized as delivery systems in the treatment of diabetes and AgNPs studied to enhance the healing of a delayed diabetic wound.43,44 The adaptability of silver NPs and data obtained by this research could be applied either on their own as it has been observed to enhance enzyme efficiency or by being functionalized to various anti-diabetic agents including insulin, thereby yielding its potential two-fold.45
Conclusion and Recommendations
TbHK and hGCK were over-expressed with a 6 his-tag in E. coli BL21 (DE3) cells containing the pRARE2 plasmid. TbHK and hGCK were characterized for their thermal and pH stability. AgNPs selectively inhibited TbHK over hGCK. TbHK showed a non-competitive inhibition with a significant decrease in Vmax and kcat/Km, respectively. hGCK showed no quantifiable effect of the nanoparticles. The significantly selective inhibitory effects of AgNPs observed between TbHK and hGCK may be helpful in the development of unique anti-trypanosomal drugs.
Abbreviations
HAT, Human African Trypanosomiasis; Tb, Trypanosoma brucei; BSF, Blood stream form; TbHK, Trypanosoma brucei hexokinase; hGCK, Human glucokinase; WHO, World Health Organisation; HIV-1, Human immunodeficiency virus-1; AgNPs, Silver nanoparticles; HsGCK, Human isoform Glucokinase; PCR, Polymerase Chain Reaction; dNTP, Deoxynucleotide triphosphate; DNA, Deoxyribonucleic acid; EDTA, Ethylenediamine tetra acetic acid; NaCl, Sodium Chloride; KCl, Potassium Chloride; MgCl2, Magnesium Chloride; RbCl, Rubidium Chloride; CaCl2, Calcium Chloride; MnCl2, Manganese Chloride; SOB, Super Optimal Broth; IPTG, isopropyl-β-D-thiogalactoside; KH2PO4, Potassium dihydrogen phosphate; Na2 HPO4, di-Sodium hydrogen phosphate; FPLC, Fast Protein liquid Chromatography; NAD, Nicotine adenine dinucleotide; MgCl2, Magnesium Chloride; HK, Hexokinase; BCA, Bicinchoninic assay; SDS-PAGE, Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis; IMAC, Immobilized Metal Chelate Affinity Chromatography; SEC, Size Exclusion Chromatography; MOPS, 3-(N-morpholino) propanesulfonic acid; CH3COOK, Potassium Acetate.
Available Data
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethical Approval and Consent to Participation
This research project was approved by the Rhodes University research department.
Consent for Publication
The authors have all consented to the publication of this research.
Acknowledgments
The authors gratefully acknowledge that this research was funded by the South African Medical Research Council (MRC).
Author Contributions
All authors made a significant contribution to the work reported, including conception, study design, execution and acquisition of data, analysis, and interpretation. All authors took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by the South African Medical Research Council (MRC).
Disclosure
The study has been presented as a thesis and interim finding are available on Rhodes University repository. The authors declare that they have no competing interests.
References
1. Truc P, Lando A, Penchenier L, et al. Human African trypanosomiasis in Angola: clinical observations, treatment, and use of PCR for stage determination of early stage of the disease. Trans R Soc Trop Med Hyg. 2012;106(1):10–14.
2. Simarro PP, Jannin J, Cattand P. Eliminating human African trypanosomiasis: where do we stand and what comes next. PLoS Med. 2008;5(2):e55. doi:10.1371/journal.pmed.0050055
3. Koagne TL, M’eyi MP, Kamkuimo RG, et al. Transmission of human African trypanosomiasis in the Komo-Mondah focus, Gabon. Pan Aft. 2011;8:36.
4. Baker N, de Koning HP, Mäser P, et al. Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol. 2013;29(3):110–118. doi:10.1016/j.pt.2012.12.005
5. Albert MA, Haanstra JR, Hannaert V, et al. Experimental and in silico analyses of glycolytic flux control in bloodstream form trypanosoma brucei. J. Biol. Chem. 2005;280(5):28306–28315. doi:10.1074/jbc.M502403200
6. Lyda T. Exploring Trypanosoma Brucei Hexokinase Biology, Localization and Inhibition Studies, Graduate School of Clemson University. [DPhil Thesis]; 2009. Available from www.etd.lib.Clemson.edu/document/1263396314/lyda_clemson/0050d/10416.pdf. Accessed May 28, 2012.
7. Chambers JW, Fowler ML. The antitrypanosomal agents lonidamine inhibit trypanosoma brucei hexokinase 1. Mol Biol Para. 2008;158:202–207. doi:10.1016/j.molbiopara.2007.12.013
8. Joice AC, Harris MT, Kahney EW. Exploring the mode of action of ebselen in Trypanosoma brucei hexokinase inhibition. Int J Parasitol. 2013;3:154–160. doi:10.1016/j.ijpddr.2013.08.002
9. Sadanandan N. Nanomedicine-The basics. West London Med J. 2011;3(3):11–14.
10. Driskell JD, Jones CA, Tompkins SM, et al. One-step assay for detecting influenza virus using dynamic light scattering and gold nanoparticles. Analyst. 2011;136:3083.
11. Bonanni A, Pividori MI, Valle M. Impedimetric detection of influenza A (H1N1) DNA sequence using carbon nanotubes platform and gold nanoparticle amplification. Carb Nano. 2010;135:1765–1772.
12. Samra Q, Ahmad S, Javeid M, et al. Anticancer medicine (Doxorubicin and Methotrexate) conjugated with magnetic nanoparticles for targeting drug delivery through iron. Prep. Biochem. Biotech. 2013;43(8):781797. doi:10.1080/10826068.2013.782042
13. Block O, Mitra A, Novotny L, et al. A rapid label-free method for quantitation of human immunodeficiency virus type-1 particles by nanospectroscopy. J. of Virol. Meth. 2012;182(1–2):70–75. doi:10.1016/j.jviromet.2012.03.012
14. Nguyen KT. Photothermal Therapy and Nanomaterials. J Bioeng Biomed Sci. 2012;2(4):e1122. doi:10.4172/2155-9538.1000e112
15. Khan M, Karuppiah P, Alkhathlan HZ, et al. Green synthesis of silver nanoparticles using Juniperus procera extract: their characterization, and biological activity. Crystals. 2022;12:420. doi:10.3390/cryst12030420
16. Asimuddin M, Mohamed RS, Fathima N, et al. Study of antibacterial properties of Ziziphus mauritiana based green synthesized silver nanoparticles against various bacterial strains. Sustain. 2020;12:1484. doi:10.3390/su12041484
17. Lara HH, Ayala-Nuñez NV, Ixtepan-Turrent L, et al. Mode of antiviral action of silver nanoparticles against HIV-1. J Nanobiotech. 2010;l8:1–10. doi:10.1186/1477-3155-8-1
18. Xiang D, Chen Q, Pang L, et al. Inhibitory effects of silver nanoparticles on H1N1 influenza A virus in vitro. J Viro l Meth. 2011;178(1–2):137142.
19. Sennuga AT. Biological Synthesis of Metallic Nanoparticles and their Interactions with Various Biomedical Targets. [PhD Thesis]. Rhodes. Available from; http://eprints.ru.ac.za/3049/. Accessed August 15, 2012.
20. Adeyemi OS, Whiteley CG. Interaction of metal nanoparticles with recombinant arginine kinase from Trypanosoma brucei; thermodynamic and spectrophotometric evaluation. J Biochimica Biophys Acta-Gen Sub. 2013;1840(1):701–706. doi:10.1016/j.bbagen.2013.10.038
21. De Moor W, Van Marwijk J, Wilhelmi BS, et al. Interaction of silver nanoparticles with triosephosphate isomerase from human and malarial parasite (Plasmodium falciparum): a comparative study. J Biomed Nanotechnol. 2015;11(6):1071–1079. doi:10.1166/jbn.2015.2003
22. Padayachee ER, Arowolo A, Whiteley CG. Nanomedicine: action of metal nanoparticles on neuronal nitric oxide synthase-fluorometric analysis on the mechanism for fibrillogenesis. J Neurochem Res. 2013;39:194–201. doi:10.1007/s11064-013-1206-x
23. Jayachandran P, Ilango S, Suseela V, et al. Green synthesized silver nanoparticle-loaded liposome-based nanoarchitectonics for cancer management: in vitro drug release analysis. Biomed. 2023;11:217.
24. Dykman LA, Khlebtsov NG. Gold nanoparticles in Biology and Medicine. Rec Adv Prosp Europe. 2011;3(9):34–55.
25. Liu C, Yang X, Yuan H, et al. Preparation of silver nanoparticle and its application to the determination of ct-DNA. Sensors. 2013;2013:708718.
26. Shen W, Feng L, Feng H, et al. Ultrafine silver (II) oxide particles decorated porous ceramic composites for water treatment. J of Chem. Eng. 2011;175:592–599. doi:10.1016/j.cej.2011.09.121
27. Johannessen CM, Boehm JS, Kim SY, et al. COT/MAP3K8 drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nat. 2010;468(7326):968–972. doi:10.1038/nature09627
28. Pal A, Shah S, Devi S. Microwave-assisted synthesis of silver nanoparticles using ethanol as a reducing agent. Mat Chem Phys. 2009;114:30–532. doi:10.1016/j.matchemphys.2008.11.056
29. Hanahan D, Harbor CS. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi:10.1016/S0022-2836(83)80284-8
30. Singh M, Singh S, Prasad S¸et al. Nanotechnology in medicine and antibacterial effect of silver nanoparticles. J Nano Biostr. 2009;3(3):115–122.
31. Hsu SL, Chung WU, Tarng R. Preparation of silver nanoparticle with different particle sizes for low- temperature sintering. Int Conf on Nanotech and Biosens. 2011;2:55–58.
32. Tiedge M, Richter T, Lenzen S. Importance of cysteine residues for the stability and catalytic activity of human pancreatic beta cell glucokinase. Arch Biochem Biophys. 2000;375(2):251–260. doi:10.1006/abbi.1999.1666
33. Bigon E, Boarato E, Bruni A, et al. Pharmacological effects of phosphatidylserine liposomes: regulation of glycolysis and energy level in brain. Br. J. Pharmacol. 1979;(1979(66):167–174. doi:10.1111/j.1476-5381.1979.tb13661.x
34. Odika IE, Asuzu IU, Anika SM. The effects of hyperosmolar agents lithium chloride and sucrose on the brain concentration of diminazene aceturate in rats. Acta Trop. 1995;60:119–125.
35. Troster SD, Muller U, Kreuter J. Modification of the body distribution of poly(methyl methacrylate) nanoparticles in rats by coating with surfactants, Int. J Pharm. 1990;61:85–100.
36. Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47:65–81. doi:10.1016/S0169-409X(00)00122-8
37. Olbrich C, Gessner A, Schröder W, et al. Lipid-drug conjugate nanoparticles of the hydrophilic drug diminazene-cytotoxicity testing and mouse serum adsorption. J Control Release. 2004;96(3):425–435. doi:10.1016/j.jconrel.2004.02.024
38. Croft SL. Pharmacological approaches to antitrypanosomal chemotherapy. Memórias Do Instituto Oswaldo Cruz. 1999;94(2):215–220. doi:10.1590/S0074-02761999000200017
39. Byrne MM, Sturis J, Clément K, et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest. 1994;93(3):1120–1130. doi:10.1172/JCI117064
40. Matschinsky FM, Porte D. Glucokinase activators (GKAs) promise a new pharmacotherapy for diabetics. F1000 med Rep. 2010;2::12–16. doi:10.3410/M2-43
41. Angadi KK, Gundampati RK, Jagannadham MV, et al. Molecular docking studies of guggultetrol from Nymphaea pubescens with target glucokinase (GK) related to type-II Diabetes. Sci. 2013;3(2):127–131.
42. Liu C, Yang X, Yuan H, et al. Preparation of silver nanoparticle and its application to the determination of ct-DNA. Sensors. 2007;2007:708–718.
43. Damgé C, Maincent P, Ubrich N. Oral delivery of insulin associated to polymeric NPs in diabetic rats. J Cont Rel. 2007;117(2):163–170. doi:10.1016/j.jconrel.2006.10.023
44. Chalasani KB, Russell-Jones GJ, Jain AK, et al. Effective oral delivery of insulin in animal models using vitamin B12-coated dextran nanoparticles. J Control Release. 2007;122(2):141–150. doi:10.1016/j.jconrel.2007.05.019
45. Mishra M, Kumar H, Tripathi K. Diabetic delayed wound healing and the role of silver. J of Nano. 2008;3(2):49–54.



