Short-Term Intravenous Administration of Carbon Nano-Onions is Non-Toxic in Female Mice

Introduction
Carbon nanomaterials (CNMs) are a family of materials composed primarily of carbon atoms arranged in a unique structure on a nanoscale. Since the discovery of C60 fullerene in 1985, the remarkable geometries, physical and chemical properties of CNMs have drawn attention for their potential use as a drug delivery system and diagnostic agent.1 Carbon nanotubes (CNTs) are the most extensively studied CNMs. Single-walled carbon nanotubes (SWCNTs) are cylindrical tubes made up of rolled-up sheets of graphene, whereas multi-walled carbon nanotubes (MWCNTs) consist of multiple layers of concentric SWCNTs, typically a few nanometers in diameter and up to several microns in length.2,3 This unique nanoscale structure offers CNTs superior properties suitable for drug delivery, including high drug loading capacity4 and excellent cellular uptake.5 However, concerns have emerged regarding the toxicity of CNTs as numerous toxicological studies have identified that CNTs can elicit a pathogenic response, including hepatotoxicity,6 inhibition of cytochrome P450s (CYP450s),7 nephrotoxicity,8 pulmonary fibrosis9 and immunotoxicity.10 Furthermore, CNTs have a similar structure to asbestos fibers, and MWCNT-7 (long, rigid CNT) has been classified as possibly carcinogenic to humans (Group 2B)11 following studies showing that intraperitoneal injection of MWCNTs caused mesothelioma in rodents.12–14 The definite cause of CNT toxicity is not fully understood, but the accumulation of CNTs (primarily in the liver, spleen and lungs) and subsequent generation of oxidative stress has been attributed as one of the key mechanisms of CNT toxicity.15 However, functionalization of SWCNTs with ZnO-Ag and ZnO-Au has shown bactericidal activity16 and developing a polyethylene glycol (PEG)-polyethyleneimine biocompatible copolymer for SWCNTs has shown cytotoxicity toward breast cancer cells.17 Thus, work on improving the biocompatibility of CNTs continues along with continued research on other carbon nanomaterials.
Carbon nano-onions (CNOs) are multi-layered fullerenes composed of concentric graphitic layers arranged in a quasi-spherical shape resembling an onion.18,19 As a member of CNMs, CNOs have physical, chemical, electrical, thermal and mechanical properties comparable to those of CNTs. A major difference is their aspect ratio, as CNTs have a long, rigid or tangling characteristic (high aspect ratio) compared to the sphere-like geometry of CNOs.20 Considering the low aspect ratio of CNOs, in vivo delivery may be more dynamically feasible. Despite their discovery nearly simultaneously with CNTs,18 CNOs have only recently received more attention in research. There is growing evidence that CNOs can be used as a delivery vehicle across the blood-brain barrier,21 intracellular delivery of glycopeptides and proteins,22 amorphous drug delivery,23 targeting cancer cells that overexpress CD44 receptors24 and achieving controlled drug release through stimuli-responsive drug delivery systems.25–27
A major challenge in the application of CNOs, as with other CNMs, is their hydrophobicity. A stable aqueous dispersion of CNOs is essential for biological applications. In order to enhance the dispersibility of CNOs, surface functionalization is a strategy used to introduce functional groups onto the CNO. Generally, an oxidation reaction is the most common covalent approach, which involves the introduction of oxygen-containing functional groups (eg carboxyl groups) on the pristine CNOs (p-CNOs) to yield oxidized CNOs (oxi-CNOs).28 This can be achieved through the treatment of CNOs with a strong oxidizing agent, such as nitric acid under reflux conditions.28 Functionalized oxi-CNOs have been shown to have excellent dispersibility and stability in aqueous and biological media.29,30
In order to be used for drug delivery or diagnostic applications, the toxicological profile of nanomaterials must be fully characterized and understood. In vitro and in vivo toxicological investigations are therefore essential prior to the practical use of CNOs. In general, the toxicity of CNOs can be affected by the synthesis methods, as these can lead to nanoparticles with differences in terms of purity, morphology and physicochemical properties. To date, in vitro studies that have reported cytotoxicity are from large CNOs (30 nm) that were synthesized by underwater arc discharge,31,32 while small CNOs (5 nm) derived from thermal annealing of nanodiamonds showed biocompatibility.22,28,29,33,34 Although in vivo evaluation of CNO toxicity is limited, small CNOs have been reported as biocompatible in freshwater polyps (Hydra vulgaris)35 and zebrafish (Danio rerio).36 Since there are no toxicological studies that have examined the systemic effects of CNOs, this study aimed to investigate the organ accumulation and whole animal toxicity of CNOs following intravenous administration in female BALB/c mice.
Materials and Methods
Materials
Acetylacetone, ammonium acetate, barium hydroxide, erythromycin, formaldehyde 37% solution, ketoconazole, 2-mercaptoethanol, paraformaldehyde, potassium chloride, potassium phosphate dibasic, sodium dodecyl sulfate, sucrose, Trizma base, Trizma hydrochloride, Tween 20 and zinc sulfate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from ECP Labchem (Auckland, New Zealand). Ethylenediamine tetra-acetic acid (EDTA) disodium salt dihydrate was purchased from BDH Laboratory Supplies (Poole, England, UK). Malonaldehyde bis (diethyl acetal) (CAS no. 122–31-6) and 2-thiobarbituric acid (TBA) were purchased from AK Scientific (Union City, CA, USA). Potassium phosphate monobasic was purchased from Fisher Scientific (Loughborough, UK). Sodium dithionite was purchased from Merck (Darmstadt, Germany). ß-nicotinamide adenine dinucleotide 2’-phosphate reduced tetrasodium salt (NADPH) (CAS no. 2646–71-1) was purchased from Roche Diagnostic (Mannheim, Germany). Bovine serum albumin (BSA) fraction V (cat no. 30060–487) was purchased from Thermo Fisher Scientific (Auckland, New Zealand). Creatinine colorimetric assay kit and superoxide dismutase (SOD) colorimetric assay kit were purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Alanine aminotransferase (ALT) reagent, Pierce bicinchoninic acid (BCA) protein assay kit and SuperSignal West Pico PLUS chemiluminescent substrate were purchased from Thermo Fisher Scientific (Rockford, IL, USA). Eosin Y, Gill II hematoxylin and optimum cutting temperature (OCT) compound were purchased from Leica Biosystems (Richmond, IL USA). Dibutylphthalate polystyrene xylene (DPX) mounting medium was purchased from Scharlab (Barcelona, Spain). Trichloroacetic acid and xylene were purchased from VWR Chemicals (Solon, OH, USA). 30% Acrylamide/bis-acrylamide solution, 29:1 and Precision Plus dual color protein standard (cat no. 1610374) were purchased from Bio-Rad Laboratories (Hercules, CA, USA). Polyvinylidene fluoride (PVDF) membrane (Immobilon-P, Millipore, cat no. IPVH00010) was purchased from Merck Millipore (Carrigtwohill, Co. Cork, Ireland). Granulated skimmed milk powder was purchased from Prontofoods (Montichiari, BS, Italy). CL-XPosure film and rabbit polyclonal CYP3A4 primary antibody (Invitrogen, cat no. PA5-14896) were purchased from Thermo Fisher Scientific (Rockford, IL, USA). Mouse monoclonal GAPDH primary antibody (Sigma-Aldrich, cat no. G8795), horseradish peroxidase-conjugated secondary antibodies, goat polyclonal anti-rabbit (Calbiochem, cat no. 401353) and goat polyclonal anti-mouse (Calbiochem, cat no. 401253) were purchased from Merck (Darmstadt, Germany). Pooled human liver microsomes (Gibco, cat no. HMMCPL, lot no. PL050F-C, 50 donors) were purchased from Thermo Fisher Scientific (Frederick, MD, USA). Ultrapure water (Type I water, 18.2 MΩ.cm resistivity, ≤5 ppb total organic carbon (TOC)) used in the study was prepared from Thermo Scientific Barnstead GenPure Pro water purification systems (Thermo Fisher Scientific, Waltham, MA, USA). Oxi-CNOs (dry powder form) were supplied by Professor Silvia Giordani (Dublin City University, Dublin, Ireland) and were characterized by a range of analytical techniques.29 These techniques included Raman spectroscopy, thermogravimetric analysis, X-ray photoelectron spectroscopy and high-resolution transmission electron microscopy (HR-TEM). p-CNOs were synthesized through thermal annealing of detonation nanodiamonds and functionalized by oxidation using nitric acid under reflux conditions to yield oxi-CNOs, as previously described.29 All other chemicals/reagents were the highest grade commercially available.
Methods
Preparation of Oxi-CNO Dispersions
Glass vials and dispersant (50% w/v PEG-200 in ultrapure water) were autoclaved before use. Subsequently, oxi-CNOs were weighed into sterile glass vials and sterile dispersant (50% w/v PEG-200 in ultrapure water) was added to achieve final concentrations of 1.25, 2.50 and 5.00 mg/mL. Oxi-CNOs were dispersed by ultrasonication for 15 min at 37 kHz and 100% intensity using an ultrasonic water bath (Elmasonic P30H, Elma Schmidbauer) and characterized by dynamic light scattering (DLS) before use. Size characterization methods are in the Supplementary Methods Section and DLS results are shown in Supplementary Figure S1 and Table S1. Two different dispersion media were used: 50% w/v PEG-200 was used for intravenous administration, while ultrapure water was the suitable dispersion medium for hepatic microsome experiments.
Animals
Female BALB/c mice (7–9 weeks old) were purchased from Hercus Taieri Resource Unit (Dunedin, New Zealand). Mice were housed in polycarbonate cages with corn cob bedding, wood shavings, and were given ad libitum access to food and water under laboratory conditions (21 ± 1°C, 55 ± 5% humidity and 12:12 h light-dark cycle) at an approved animal facility. All mice were acclimatized for 3 days before experimentation. All experimental procedures were approved by the University of Otago Animal Ethics Committee (AUP-18-224).
CNO and Drug Administration
Mice (n=5 per group) were randomly allocated to the following treatment groups: oxi-CNOs (125, 250 or 500 µg, iv) given as a single dose or 3 doses every 4 days; vehicle control (50% w/v PEG-200, iv) given as a single dose or 3 doses every 4 days. All were administered via the tail vein. The mice were monitored for 7 days after the single dose, while mice receiving three doses of oxi-CNOs were monitored for a total of 12 days. Time-course studies were also conducted where mice were administered with a single dose of oxi-CNOs (500 µg, iv) or vehicle control (50% w/v PEG-200, iv) and euthanized at 6, 12, 24 and 48 h. The mice used for positive controls were treated with either carbamazepine (200 mg/kg, po, daily for 5 days),37 dexamethasone (50 mg/kg, po, daily for 4 days)38 or D-galactose (300 mg/kg, ip, daily for 7 days).39
Physiological Parameters and Overall Animal Health
The general behavior and body weight of the mice were monitored daily throughout the various observation windows. Mice were euthanized by CO2 inhalation either 24 h after the last dose or at the time indicated. Blood was collected immediately from the inferior vena cava and stored on ice. Mice were then perfused through the heart with 5 mL of phosphate-buffered saline (PBS) and a full necropsy was performed in which major organs, including the liver, spleen, lungs and kidneys were collected and weighed. Organ weight is expressed as a percentage of body weight.
Measurement of ALT Activity and Creatinine Level
Blood collected was centrifuged at 5000 rpm for 5 min at 4°C and the resulting plasma supernatant was stored at −20°C until analysis. Plasma ALT activity and creatinine level were measured with the ALT colorimetric assay kit and creatinine colorimetric assay kit respectively,40 according to the manufacturer’s instructions. The results for ALT activity are expressed as international units per liter (IU/L) and the results for creatinine level are expressed as mg/dL.
Histological Evaluation
Organs of interest were trimmed to the appropriate sizes for histological evaluation and the remaining portion was used for the preparation of microsomes and homogenates. Excised tissues were fixed in 4% w/v paraformaldehyde for 24 h before being placed in 30% w/v sucrose (in PBS) for a further 24 h. The tissue specimens were then snap-frozen in OCT compound with liquid nitrogen and stored at −20°C until analysis. OCT-embedded liver and kidney tissues were cut into 12 µm sections while spleen and lung tissues were cut into 6 µm sections using a cryostat (Thermo Scientific HM 525, Thermo Fisher Scientific). The tissue sections were mounted on microscope slides, stained with hematoxylin and eosin (H&E), cleared in xylene, mounted with DPX and coverslipped. Stained slides were scanned using a digital pathology scanner (Aperio ScanScope CS, Leica Biosystems) at 20× (image resolution: 0.5 μm/pixel) and 40x (image resolution: 0.25 μm/pixel) magnification. Images were acquired using Aperio ImageScope digital pathology software (version 12.4.3.5008, Leica Biosystems) and the dimension of the images was fixed at 1716×942 pixels. Blinded analysis of the 20× scanned images was performed with ImageJ software (version 1.53k).
Preparation of Liver Microsomes and Organ Homogenates
Liver microsomes were prepared by differential centrifugation, as previously described38 and stored at −80°C until analysis. Spleen homogenates were prepared as previously described.41 Briefly, spleens were homogenized in ice-cold 1 mM Tris-HCl buffer containing 0.1 mM EDTA-2Na, 0.8% NaCl (pH 7.4) to yield 10% w/v homogenates. Homogenates were centrifuged at 2000 rpm for 8 min at 4°C. The supernatant was collected and stored at −80°C until analysis. All other organs of interest (lungs and kidneys) were placed in PBS (pH 7.4) and homogenized. Homogenates were centrifuged at 10,000 rpm for 10 min at 4°C.42 The supernatant was collected and stored at −80°C until analysis. Protein concentration of the liver microsomes and organ homogenates was determined with the BCA protein assay kit according to the manufacturer’s instructions using a BSA standard curve.43
Quantification of Total Hepatic CYP450
Total hepatic CYP450 was quantified by measuring the absorbance spectra of reduced CYP450, as previously described.44,45 Liver microsomes were diluted to a final protein concentration of 1 mg/mL using Tris buffer (1 M, pH 7.4) in a cuvette. The samples were reduced by adding a spatula tip of solid sodium dithionite (~1 mg) and were gently mixed by five inversions (covered with parafilm). Carbon monoxide was bubbled into the samples at a rate of 1 bubble per second for 30s. The absorbance at 450 nm and 480 nm was measured using a visible spectrophotometer (Thermo Scientific Genesys 30, Thermo Fisher Scientific). Quantification of total hepatic CYP450 was obtained by calculating the absorbance difference between 450 nm and 480 nm, with an extinction coefficient of 91 mM−1cm−1.45 The results are expressed as nmol/mg protein.
Hepatic CYP3A Catalytic Activity
Hepatic CYP3A catalytic activity was determined by erythromycin N-demethylation, through the quantification of formaldehyde using the Nash method,46 as previously described with slight modification.47 The reaction was carried out in a test tube and the reaction mixture consisted of the following: liver microsomes (1 mg of total protein), erythromycin buffer (0.1 M potassium phosphate buffer, 0.1 mM EDTA and 0.4 mM erythromycin, pH 7.4) and potassium phosphate buffer (0.1 M, pH 7.4) in a final volume of 1 mL. The reaction was initiated by the addition of 50 mM NADPH and incubated at 37°C in a shaking water bath for 30 min. The reaction was terminated by the addition of 15% w/v zinc sulfate solution and incubated at room temperature for 5 min. The samples were further incubated for another 5 min at room temperature after the addition of saturated barium hydroxide solution. The samples were then centrifuged at 2000 rpm for 10 min at 4°C. The resulting 0.83 mL supernatant was reacted with an equal volume of Nash reagent (30% w/v ammonium acetate, 0.4% v/v acetylacetone) at 60°C in a shaking water bath for 30 min. The samples were centrifuged at 2000 rpm for 10 min at 4°C and the absorbance of the supernatant was measured at 415 nm using a visible spectrophotometer (Thermo Scientific Genesys 30, Thermo Fisher Scientific). The catalytic activity of CYP3A was determined from a standard curve of formaldehyde. The results are expressed as nmol/mg protein/min.
Western Blotting Analysis of CYP3A Proteins
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as previously described.48 Briefly, liver microsomes were solubilized in Laemmli buffer (4 x) at a total protein concentration of 1 mg/mL and heated at 95°C for 5 min. 20 µg of protein was then loaded onto the wells of a 10% polyacrylamide gel and the Precision Plus dual color protein standard was used as the molecular weight marker. Gel electrophoresis was run at 80 V (~20 min) to allow protein stacking and then increased to 120 V until the bromophenol blue marker reached the bottom of the gel (~1.5 h), using a Mini-Protean 3 Cell (Bio-Rad Laboratories). Western blotting analysis of CYP3A protein was performed as previously described.49 The proteins were transferred from the gel to the PVDF membrane at 100 V for 100 min, via a wet transfer system (Mini Trans-Blot Cell, Bio-Rad Laboratories). The membranes were washed (5 min) with Tris-buffered saline (TBS, pH 7.4) and were blocked (1 h, room temperature) in a blocking buffer. The blocking buffer contained 5% w/v BSA in TBST (TBS with 0.05% Tween 20, pH 7.4). The membranes were subsequently incubated (20 h, 4°C) with the respective primary antibodies diluted in a blocking buffer. The primary antibodies used were CYP3A (1:550) and GAPDH (1:4500). At the end of the incubation period, the membranes were rinsed (3 x) with ultrapure water and washed (4 x 5 min) with TBST. Chemiluminescence detection was achieved by using horseradish peroxidase-conjugated secondary antibodies (1:3000). Goat anti-rabbit and goat anti-mouse antibodies were used for CYP3A and GAPDH, respectively. The membranes were incubated (1 h, room temperature) with the respective secondary antibodies in TBST, containing 5% w/v of non-fat dry milk. The membranes were then washed (4 x 5 min) with TBST before the addition of SuperSignal West Pico PLUS chemiluminescent substrate and visualized using CL-XPosure film. Images were scanned with an imaging densitometer (GS-710, Bio-Rad Laboratories) and analyzed with Quality One software (version 4.6.7, Bio-Rad Laboratories). The signal intensity of CYP3A was normalized to the housekeeping protein, GAPDH and the results are expressed as a percentage of untreated control.
In vitro Inhibition of Hepatic CYP3A Catalytic Activity
Hepatic CYP3A catalytic activity was determined following in vitro incubation of oxi-CNOs with liver microsomes from untreated male BALB/c mice and pooled human donors. The experiment was carried out according to the procedure described in the hepatic CYP3A catalytic activity section. However, the test compounds, 50, 100 and 200 µg/mL of oxi-CNOs dispersed in ultrapure water (see Supplementary Figure S1 and Table S1 for size characterization), were also added to the reaction mixture containing untreated mouse or human liver microsomes. Ultrapure water was used as a dispersion medium control. Ketoconazole (2.5 µM) was used as a positive control50 and DMSO was used as a solvent control for ketoconazole.
Measurement of Lipid Peroxidation
Malondialdehyde (MDA) level was quantified to determine the presence of lipid peroxidation in liver microsomes and organ homogenates from oxi-CNO-treated mice, as previously described.51 Briefly, samples (1 mg/mL protein) were added to a test tube containing 10% w/v trichloroacetic acid. The samples were incubated on ice for 15 min before centrifugation at 2000 rpm for 10 min at 4°C. The resulting 500 µL of the supernatant was reacted with an equal volume of 0.67% w/v TBA by heating in a boiling water bath (~100°C) for 10 min. The samples were cooled in an ice water bath for 5 min and the absorbance was measured at 532 nm using a visible spectrophotometer (Thermo Scientific Genesys 30, Thermo Fisher Scientific). The amount of MDA was determined from a standard curve of MDA. The MDA standard was prepared using malonaldehyde bis (diethyl acetal). The results are expressed as nmol/mg protein.
Measurement of SOD Activity
SOD activity in liver microsomes and organ homogenates (1 mg/mL protein) was measured with the SOD colorimetric assay kit52 according to the manufacturer’s instructions. The results are expressed as units per milligram of protein (U/mg protein). One unit of SOD is defined as the amount of enzyme needed to exhibit 50% dismutation of the superoxide radical.
Statistical Analysis
All data are presented as the mean ± SEM unless otherwise stated. The number of replicates performed is indicated in each table/figure legend, where applicable. For all experiments with one factor, statistical significance was determined by a one-way analysis of variance (ANOVA). For the analysis of two factors (ex. aggregate counts in tissue and dose), statistical significance was determined by a two-way ANOVA. All significant ANOVAs were coupled to a Tukey’s multiple comparisons post-hoc test in which p<0.05 was required for a statistically significant difference. All statistical analyses were carried out using GraphPad Prism software (version 9.3.1, GraphPad Software).
Results
Assessment of the General Toxicity of CNOs
In order to determine the safety of CNOs for drug delivery, the mice were intravenously administered a single dose of oxi-CNOs (125, 250 or 500 µg) or vehicle control (50% w/v PEG-200). The administration of oxi-CNOs was well tolerated by mice over 7 days of monitoring, as there were no significant changes to mouse behavior, body weight and organ weight for all the treatment groups compared to untreated control (Supplementary Table S2). Additionally, oxi-CNOs did not cause hepatotoxicity, as plasma ALT activity was consistently within the normal range of 0–80 IU/L (Table 1). Likewise, there was no evidence of nephrotoxicity, as all the mice had normal plasma creatinine that was below the threshold of 2 mg/dL (Table 1). Hence, the dosing regimen was well-tolerated by the mice and repeated administration of oxi-CNOs was then studied. Mice receiving three doses of oxi-CNO (125, 250 or 500 µg) or vehicle control (50% w/v PEG-200) showed significant body weight loss (<4%) over 12 days compared to untreated control (p<0.05). Specifically, while untreated mice gained an average of 1.16 g over 12 days, mice administered three doses of 250 and 500 µg of oxi-CNO lost an average of 0.68 and 0.62 g, respectively (Supplementary Table S2). However, the weight of the liver, spleen, lungs and kidneys of the mice showed no significant changes compared to untreated mice (Supplementary Table S2). Moreover, there was no evidence of hepatotoxicity or nephrotoxicity, as plasma ALT activity and creatinine levels were consistently within the normal range (Table 1).
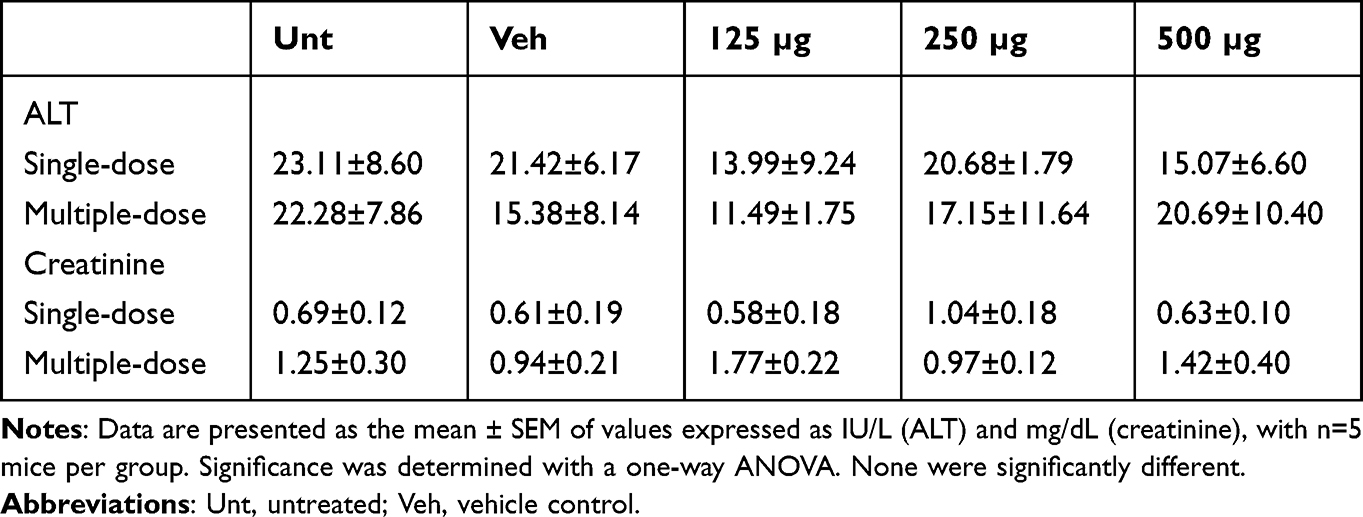 |
Table 1 Plasma ALT Activity and Creatinine Levels Following Treatment with Oxi-CNOs |
Histological Evaluation
H&E staining was used to provide a high-contrast visualization to identify areas of the tissues containing CNO aggregates which are black in color. A single dose of oxi-CNOs resulted in an accumulation of CNO aggregates in the liver, spleen and lungs (Supplementary Figure S2). Likewise, CNO aggregates also accumulated in the liver, spleen, lungs and kidneys in mice receiving three doses of oxi-CNOs (Figure 1A). However, no accumulation of black aggregates was observed in the tissues of untreated or vehicle control mice (Figure 1A). Quantification of the black aggregates showed a significant dose-dependent accumulation in the liver, spleen, lungs and kidneys, as the aggregate number was significantly increased in each organ with an increase in the oxi-CNO dose (Figure 1B and C). At the highest dose, there were significantly more aggregates in the spleen compared to all other organs. Specifically, in the mice treated with three doses of 500 µg oxi-CNOs, the spleen showed the highest mean aggregate counts (1101 ± 57), followed by the liver (536 ± 34) and lung (240 ± 20) (Figure 1B). The kidneys showed the lowest CNO accumulation, specifically the mean aggregate counts in 125, 250 and 500 µg oxi-CNOs treated mice were 7 ± 2, 4 ± 1 and 3 ± 1, respectively (Figure 1C).
Assessment of CYP450
The accumulation of CNO aggregates in the organs is a potential concern. In particular, the liver is a key organ involved in the CYP450-catalyzed metabolism and detoxification of xenobiotics. Therefore, the potential impact of CNOs on CYP450 enzymatic function was studied. The total hepatic CYP450 content in the liver microsomes was measured to determine if CNOs altered the levels of CYP450. The results showed that there were no significant differences between the treatment groups compared to vehicle control, indicating that three doses of oxi-CNOs (125, 250 or 500 µg) did not alter the hepatic CYP450 content in the liver (Figure 2A). However, dexamethasone, a known inducer of total hepatic CYP450,53 showed a 1.7-fold increase in total hepatic CYP450 compared to vehicle control (p<0.0001). To confirm this finding, the catalytic activity and protein levels of CYP3A were examined. CYP3A was chosen as it is the most abundant CYP subfamily and has a broad substrate specificity involved in the metabolism of most clinically used drugs.54,55 The results showed that there were no significant differences between the treatment groups and vehicle control, indicating that three doses of oxi-CNOs (125, 250 or 500 µg) did not affect the catalytic activity of CYP3A (Figure 2B). However, carbamazepine, a known inducer of CYP3A,37 increased CYP3A catalytic activity 3.1-fold higher compared to that of the vehicle control (p<0.0001). CYP3A protein levels were then examined to ensure that these were also unchanged. The results from Western blotting experiments showed that there were no significant changes in the CYP3A protein levels between the treatment groups compared to untreated control (Figure 2C and D). However, carbamazepine increased CYP3A protein levels 2.1-fold compared to untreated control (p<0.0001).
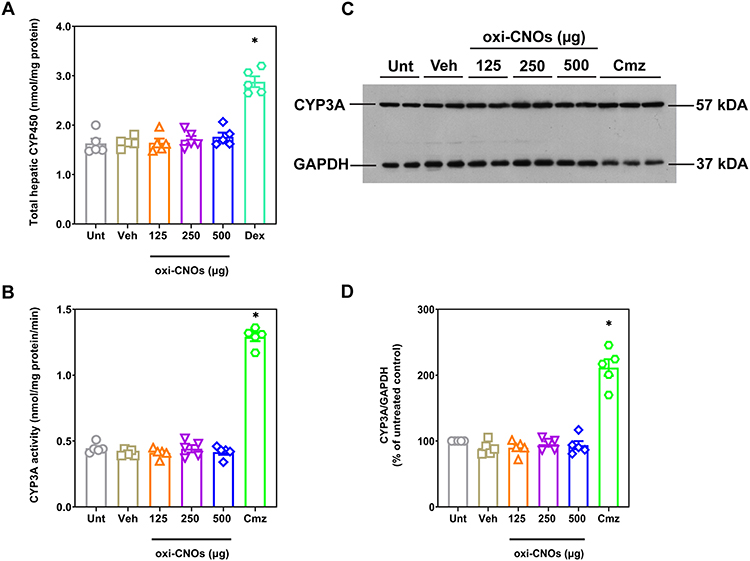 |
Figure 2 Hepatic CYP450 and CYP3A protein levels following treatment with three doses of oxi-CNOs or vehicle control (50% w/v PEG-200). Mice treated with dexamethasone (50 mg/kg, po, daily for 4 days) and carbamazepine (200 mg/kg, po, daily for 5 days) were used as positive control. (A) Total hepatic CYP450 (B) CYP3A catalytic activity. (C) Representative Western blot for CYP3A and housekeeping protein GAPDH. The positions of the molecular weight markers are indicated on the right (kDa). (D) Scanning densitometry of the CYP3A Western blots normalize to the housekeeping protein GAPDH, analyzed on 3 independent Western blots. Mean ± SEM, with n=5 mice per group. Significance was determined with a one-way ANOVA coupled with a Tukey’s post-hoc test. *Significantly increased from all other groups, p<0.0001. Abbreviations: Unt, untreated control; Veh, vehicle control; Dex, dexamethasone; Cmz, carbamazepine. |
In vitro Inhibition of Hepatic CYP3A Catalytic Activity
Previous work identified that SWCNTs were able to inhibit CYP3A in vitro through competitive binding.56 Therefore, experiments were performed to determine if CNOs could compete with erythromycin and inhibit CYP3A catalytic activity. Specifically, the catalytic activity of CYP3A in mouse liver microsomes was determined, following in vitro incubation with oxi-CNOs at 50, 100 and 200 µg/mL. Ultrapure water was used to disperse oxi-CNOs because 50% w/v PEG-200 showed a 9.7-fold increase in CYP3A activity compared to ultrapure water alone (p<0.0001). Even though 0.2% DMSO has been reported to inhibit CYP3A,57 5% DMSO did not inhibit CYP3A activity in the current study. Therefore, DMSO was used as a solvent control for ketoconazole. The results showed that there were no significant differences between the treatment groups compared to dispersion medium control (ultrapure water), indicating that CNOs did not act as competitive inhibitors of mouse CYP3A (Table 2). However, ketoconazole (2.5 µM), a known potent inhibitor58 decreased erythromycin N-demethylation by 60% compared to its solvent control. To determine if this effect was consistent across species, the experiments were also performed with human liver microsomes. The results showed that CNOs also failed to inhibit erythromycin N-demethylation in human liver microsomes (Table 2), indicating that the effects of oxi-CNOs in mice translated to humans.
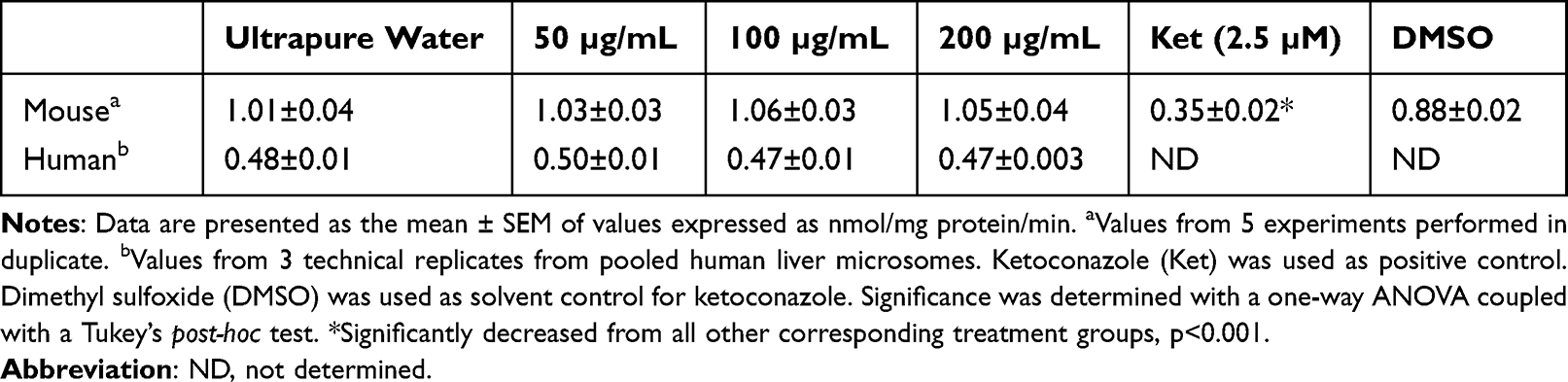 |
Table 2 Hepatic CYP3A Catalytic Activity Following in vitro Incubation with Oxi-CNOs |
Assessment of Oxidative Stress
Nanoparticle-induced toxicity is often associated with oxidative stress.59–61 Therefore, the generation of reactive oxygen species (ROS) was characterized to determine if CNOs induced oxidative stress. However, the direct detection of ROS remains elusive due to their short half-lives and high reactivity. MDA is a stable end product of lipid peroxidation induced by ROS and thus can be used as an indirect measure of oxidative stress.62 Hence, MDA was used to determine if CNO treatment induced oxidative stress in liver microsomes and organ homogenates. The results showed that there were no significant differences between the treatment groups and vehicle control in liver microsomes and spleen homogenates, indicating that three doses of oxi-CNOs (125, 250 or 500 µg) did not induce lipid peroxidation in the liver and spleen (Table 3). D-galactose, a known inducer of oxidative stress,39 showed a 2.4-fold increase in the amount of MDA in the spleen homogenates compared to vehicle control (p<0.001). However, ROS can be scavenged by antioxidants once the defense mechanism is activated.6 Therefore, the activity of SOD, the first-line defense antioxidant enzyme against ROS63 was measured and the results showed that oxi-CNOs did not alter SOD activity in the liver microsomes and spleen homogenates (Table 3). To ensure that induction of lipid peroxidation did not occur at an earlier time-point, MDA levels and SOD activity were measured across an earlier time-course (6 to 48 h). There were also no significant changes in MDA levels or SOD activity between the treatment groups and vehicle control in the liver microsomes or spleen, lung and kidney homogenates at 6 to 48 h following a single dose of 500 µg of oxi-CNOs (Table 4). Only the highest dose was used in the time-course study in order to minimize the use of animals. Since there were no changes in MDA or SOD, the experiments were not repeated at lower doses.
 |
Table 3 MDA Levels and SOD Activity in Liver Microsomes and Spleen Homogenates from Mice Treated with Three Doses of Oxi-CNOs |
 |
Table 4 Time Course of MDA Levels and SOD Activity in Liver Microsomes and Organ Homogenates from Mice Treated with a Single Dose of Oxi-CNOs (500 µg, Iv) |
Discussion
CNOs hold great potential for use as a delivery vehicle for drugs or diagnostic agents. However, systemic in vivo studies are needed to further develop their use in medical applications. In this study, we investigated the toxicity of oxi-CNOs elicited by intravenous administration in female BALB/c mice. A single administration of oxi-CNOs (125, 250 or 500 µg) over 7 days of monitoring was well tolerated by the mice. There were no changes in mouse behavior, body weight or organ weight, and neither hepatotoxicity nor nephrotoxicity was observed. A 100% survival rate has also been reported when mice were treated with 250 and 500 µg of sphere-like carbon nanocapsules and C60 fullerene over a 7-day period.64 However, the long and rigid CNTs (500 µg) elicited 18% mortality.64 Since the CNOs were well-tolerated by the mice, the effect of repeated CNO administration was examined using a dosing regimen of three intravenous injections of oxi-CNOs (125, 250 or 500 µg) over 12 days. Following this regimen, there was a significant reduction in body weight. However, there were no significant changes in mouse behavior or organ weight and there was also no evidence of hepatotoxicity or nephrotoxicity. This finding is supported by a previous study showing that male BALBcAJcl mice lost ~6% of their body weight after 10 days of tube restraint.65 Therefore, the body weight reduction (<4%) with both oxi-CNOs and vehicle control may be a result of the stress of tube restraint and multiple intravenous injections rather than the oxi-CNOs themselves.
Single administration of oxi-CNOs caused an accumulation of CNO aggregates in the spleen, liver and lung, with a trace amount of aggregates appearing in the kidneys. Following repeated administration this accumulation was dose-dependent. Furthermore, three doses of oxi-CNOs resulted in ~3.5 times higher accumulation of aggregates compared to the single dose, suggesting that there was no significant elimination of CNOs during the twelve days. The findings showing a major accumulation of CNO aggregates in the liver, spleen and lung are consistent with the general trend of high uptake of nanomaterials by the mononuclear phagocyte system (MPS).41,66–69 While urinary excretion was not measured, considering that the size of CNOs (~60 nm, Supplementary Table S1 and Figure S1) exceeded the renal filtration-size threshold of 5.5 nm,70 it is unlikely that CNOs would be renally excreted unless they were degraded beforehand. Therefore, the aggregates escaping MPS’s uptake into the kidneys were trapped in the glomerulus (Supplementary Figure S3). Notably, the results showing accumulation of CNO aggregates indicate that further investigation of the biodistribution of CNOs in mice is warranted. It is important to note that the number of aggregates quantified in this study may not fully represent the complete biodistribution. Quantification using ImageJ analysis did not allow an accurate estimation of the aggregate size and this was variable within the tissue.
CNMs are well-known for their strong thermal, chemical and mechanical durability due to their unique structural properties. These unique characteristics make them highly stable and difficult to degrade. Studies have reported that CNTs can be biodegraded by macrophages via oxidative degradation71,72 and the degradation was also confirmed by the structural defects of CNTs.73,74 CNTs were also found to be excreted through the biliary pathway, with SWCNTs detected in feces 8 h post intravenous administration.66 However, renal excretion is still the preferred route of excretion for clinically approved formulations. Biodegradation of CNMs in a living system is a complex process and biliary excretion is slow. As a consequence, a critical question concerning CNO toxicity is whether the retention of aggregates causes an impact on organ function. Therefore, the effect of CNOs on the major drug-metabolizing enzyme family, CYP450s, was assessed. The results showed that CNOs administered to mice did not alter the total hepatic CYP450 content or the catalytic activity and protein levels of CYP3A. Importantly, the total hepatic content and the catalytic activity of CYP3A for untreated mice were similar to the values previously reported.75,76 Furthermore, oxi-CNOs did not act as CYP3A competitive inhibitors in vitro.
These are important findings, as there is limited work examining CYP450s following administration of other nanoparticles. For example, zinc oxide nanoparticles (100, 300, or 600 mg/kg) administered orally to rats for 7 days significantly inhibited testosterone metabolism by CYP3A.77 In contrast, silver nanoparticles administered orally to rats (50 to 1000 mg/kg) for 2 weeks did not exhibit any changes in CYP3A activity, as measured by midazolam 1-hydroxylation, and TEM analysis of liver sections revealed that no silver particles were detected.78 A key factor that determines whether nanoparticles can affect CYP450-mediated metabolism is their ability to enter cells and exert effects. Studies have shown that CNOs accumulated in lysosomes around the nuclei of MCF-7 cells after a 24 h incubation.29 Due to the ability of CNOs to access subcellular compartments, it is likely that CNOs would come into contact with the drug-metabolizing enzymes in the endoplasmic reticulum. Additionally, normal plasma ALT activity was also observed in all the mice, indicating there was no major loss of the parenchymal or other liver cell integrity which may impair cellular uptake. However, it is important to note that when administered into the blood, the surface of CNOs would be coated with protein corona and other biomolecules, which is likely to affect cellular uptake.79 The formation of protein corona has been shown to influence the mechanisms of nanoparticle internalization and lower cell membrane adhesion of nanoparticles, resulting in a decrease in internalization efficiency.80,81 Therefore, there is a possibility that the CYP450s were not altered because the amount of CNOs reaching the subcellular compartments was too low to cause an effect. However, the in vitro results provide further support, showing that CNOs at concentrations between 50 and 200 µg/mL did not inhibit CYP3A catalytic activity.
In contrast, SWCNTs (50 to 100 µg/mL) inhibited the metabolism of testosterone (50 to 80%) in a concentration-dependent manner when studied using bactosomes expressing human CYP3A.7 Nanodiamonds, graphene oxide and graphite (3.125 to 100 µg/mL) were also found to inhibit CYP3A catalytic activity by 50 to 60% when tested with baculosomes expressing human CYP3A.82 In addition, CYP3A inhibition mediated by SWCNTs was shown to be mitigated by PEGylation and protein corona (BSA).7 Contrary to the earlier findings, however, the current in vitro study showed that CNOs without PEGylation did not affect CYP3A catalytic activity. It is known that PEGylation is a common surface modification applied to nanoparticles to reduce non-specific protein adsorption and toxicity caused by pristine surfaces.83,84 The in vitro results showing that CNOs did not affect CYP3A without the surface being modified with PEG is an advantage because it eliminates the concerns regarding toxicity that may occur in vivo due to PEG detachment. Additionally, a further in vitro study to evaluate the activity of CYPs in the presence and absence of a protein corona would characterize the surface parameters that may influence the interaction of CNOs with CYPs.
Although the amount of carbon nanomaterials taken up by specific liver cells is unknown, previous studies estimated that 70% of the injected dose of SWCNTs (100 µg, iv) was accumulated in the liver 24 h after injection.66 Furthermore, a total of 21.3% of the injected dose of SWCNTs (200 µg, iv) remained in the liver 28 days after administration.85 Therefore, the concentrations of CNOs (50 to 200 µg/mL) used for the in vitro study are realistic compared to the actual amount accumulated in the liver, as reported in the literature. Molecular dynamic simulations have identified that CNTs may inhibit enzymes via three possible mechanisms, including disruption of the active site, competitive binding and blocking access to the active site.7,56,86 A possible explanation for the absence of the alteration of CYP450s may be attributed to the weak hydrophobic interactions between CNOs and CYP450s. Inhibition of CYP450s can lead to an increase in the bioavailability of drugs or a decrease in the amount of pharmacologically active drugs in the case of prodrugs, which can result in adverse drug reactions or therapeutic failure. The absence of the alteration of CYP450s, thus, indicates that currently CNOs are generally safer than CNTs. However, improvements to CNTs continue to be made with more biocompatible functionalizations16,17 and thus the safety profile of CNTs is likely to improve.
Nanoparticle-mediated toxicity can be caused by various factors, but oxidative stress is known to be a common paradigm.59–61 An imbalance between the production of ROS and its elimination by antioxidants elicits oxidative stress.87,88 In particular, cellular and organelle membranes are vulnerable to the attack of ROS under oxidative stress due to the abundance of polyunsaturated fatty acids, resulting in lipid peroxidation.89 Therefore, lipid peroxidation was measured to determine if CNOs induced oxidative stress. Additionally, the activity of SOD, one of the major antioxidants in the cell, was also examined to determine if the antioxidant defense mechanism had been activated. SOD catalyzes the dismutation of superoxide radicals, preventing and suppressing free radical formation.63 It was found that repeated administration of CNOs did not cause lipid peroxidation or changes in SOD activity in the liver or spleen. Time-course studies also revealed no evidence of lipid peroxidation in the liver, spleen, lungs or kidneys within 6 to 48 h after a single dose of 500 µg oxi-CNO and this lack of lipid peroxidation was not regulated by changes in SOD. CNOs therefore did not induce oxidative stress. Moreover, there was no evidence that there was any hepatotoxicity or nephrotoxicity, which supports the lack of oxidative damage. These findings are also consistent with previous in vitro studies demonstrating CNO biocompatibility. p-CNOs and oxi-CNOs (30 to 300 µg/mL) were reported to be biocompatible in rat dermal fibroblasts, with almost 100% viability after a 4 h incubation.33 Likewise, oxi-CNOs (0.5 to 20 µg/mL) also showed greater than 80% viability in MCF-7 and HeLa cells after 72 h.29 In contrast, Xu et al32 found that CNOs inhibited cell viability (IC50: 44 µg/mL), caused DNA damage and induced apoptosis in human umbilical vein endothelial cells, which was attributed to ROS. The discrepancy between these results is likely due to the different methods used to synthesize CNOs. In the study of Xu et al,32 CNOs were synthesized using an arc discharge method with two graphite electrodes immersed in water, whereas studies showing their biocompatibility used nanodiamonds that had been annealed at 1650°C before being oxidized with nitric acid for 48 h.29,33 An underwater arc discharge method was initially reported to produce highly pure CNOs,90 a HR-TEM analysis, however, revealed that CNOs were contaminated with carbonaceous impurities such as polyaromatic hydrocarbons, amorphous carbon, CNTs and graphitic debris, as well as metallic impurities detected by thermogravimetric analysis.91 Although the amount of impurities in CNOs synthesized by annealing of nanodiamonds is unknown/has not been accurately analyzed (ie with a sensitive analytical method), the high temperature and acid treatment used during the synthesis process may have removed the impurities.92,93 Metallic impurity-induced oxidative stress has been identified as one of the key mechanisms causing toxicity of CNTs, as demonstrated by a reduction in viability (38%) of human keratinocyte cells after incubation with SWCNTs (30% w/w Fe, 0.24 mg/mL) for 18 h. This was evidenced by a decline in the antioxidant glutathione (48%) as well as an increase in lipid peroxidation products (46%). However, the reduction in viability was dramatically reversed by the metal chelator, deferoxamine.94 Metallic residues such as Fe, play a critical role in the generation of hydroxyl radicals through Fenton reaction, causing intracellular oxidative stress.95 Therefore, carbonaceous and metallic impurities are likely to be the cause of ROS generation and toxic effects seen in the in vitro study of CNOs synthesized by the underwater arc discharged method. On the contrary, the current study did not observe any toxicity or oxidative damage in mice. This may be due, in part, to the lack of impurities in the oxi-CNOs synthesized through the detonation of nanodiamonds and oxidation with nitric acid.
Conclusions
In this study, we investigated the safety of CNOs for their potential use as carriers for drugs and diagnostic agents. Single or repeated administration of oxi-CNOs did not affect mouse behavior or organ weight and there was also no evidence of hepatotoxicity or nephrotoxicity. However, vehicle control mice and those receiving three doses of oxi-CNO lost ~4% of their body weight, indicating that stress of chemical administration has an impact on animal weight. Although CNOs caused a dose-dependent accumulation of CNO aggregates in the organs, the accumulation of aggregates did not affect the protein level or function of CYP450s or cause oxidative stress. While additional work must be conducted to confirm long-term biocompatibility, we provide evidence for the first time that short-term intravenous administration of oxi-CNOs is non-toxic to female mice. Future research could explore a variety of surface functionalizations (eg covalent PEG) to minimize accumulation and achieve urinary excretion of CNOs. Lastly, this study has provided evidence that CNOs warrant further investigation for use in nanomedicine.
Data Sharing Statement
All data analyzed in this study are included in this article and its Supplementary Information.
Ethics Approval and Informed Consent
All animal experiments in this study were conducted in accordance with the University of Otago Code of Ethical Conduct for the Use of Animals in research, which complies with the New Zealand Animal Welfare Act 1999. All experiment procedures were approved by the University of Otago Animal Ethics Committee (AUP-18-224).
Acknowledgments
The work was supported by a University of Otago Research Grant awarded to R.J.R. and S.G. L.R.T. was supported by the University of Otago Biomedical Sciences Honors Scholarship and the Fred Fastier Summer Studentship. Y.Z.T. was supported by the University of Otago International Master’s Research Scholarship and the University of Otago Master’s Postgraduate Publishing Bursary. The authors would like to thank Abigail R. Bland for her technical assistance. Components of this paper were uploaded to the University of Otago University repository as a thesis in 2022.96
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation or in all these areas; took part in the drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Patel KD, Singh RK, Kim H-W. Carbon-based nanomaterials as an emerging platform for theranostics. Mater Horiz. 2019;6(3):434–469.
2. Iijima S, Ichihashi T. Single-shell carbon nanotubes of 1-nm diameter. Nature. 1993;363(6430):603–605. doi:10.1038/363603a0
3. Iijima S. Carbon nanotubes: past, present, and future. Physica B Condens Matter. 2002;323(1–4):1–5. doi:10.1016/S0921-4526(02)00869-4
4. Liu Z, Sun X, Nakayama-Ratchford N, Dai H. Supramolecular chemistry on water-soluble carbon nanotubes for drug loading and delivery. ACS Nano. 2007;1(1):50–56. doi:10.1021/nn700040t
5. Kang B, Li J, Chang S, et al. Subcellular tracking of drug release from carbon nanotube vehicles in living cells. Small. 2012;8(5):777–782. doi:10.1002/smll.201101714
6. Ji Z, Zhang D, Li L, et al. The hepatotoxicity of multi-walled carbon nanotubes in mice. Nanotechnology. 2009;20(44):445101. doi:10.1088/0957-4484/20/44/445101
7. El-Sayed R, Bhattacharya K, Gu Z, et al. Single-walled carbon nanotubes inhibit the cytochrome P450 enzyme, CYP3A4. Sci Rep. 2016;6(1):1–12. doi:10.1038/srep21316
8. Awogbindin IO, Maduako IC, Adedara IA, et al. Kolaviron ameliorates hepatic and renal dysfunction associated with multiwalled carbon nanotubes in rats. Environ Toxicol. 2021;36(1):67–76. doi:10.1002/tox.23011
9. Qin Y, Li S, Zhao G, et al. Long-term intravenous administration of carboxylated single-walled carbon nanotubes induces persistent accumulation in the lungs and pulmonary fibrosis via the nuclear factor-kappa B pathway. Int J Nanomedicine. 2017;12:263. doi:10.2147/IJN.S123839
10. Zhang T, Tang M, Zhang S, et al. Systemic and immunotoxicity of pristine and PEGylated multi-walled carbon nanotubes in an intravenous 28 days repeated dose toxicity study. Int J Nanomedicine. 2017;12:1539. doi:10.2147/IJN.S123345
11. Grosse Y, Loomis D, Guyton KZ, et al. Carcinogenicity of fluoro-edenite, silicon carbide fibres and whiskers, and carbon nanotubes. Lancet Oncol. 2014;15(13):1427–1428. doi:10.1016/S1470-2045(14)71109-X
12. Poland CA, Duffin R, Kinloch I, et al. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat Nanotechnol. 2008;3(7):423–428. doi:10.1038/nnano.2008.111
13. Takagi A, Hirose A, Futakuchi M, Tsuda H, Kanno J. Dose‐dependent mesothelioma induction by intraperitoneal administration of multi‐wall carbon nanotubes in p53 heterozygous mice. Cancer Sci. 2012;103(8):1440–1444. doi:10.1111/j.1349-7006.2012.02318.x
14. Takagi A, Hirose A, Nishimura T, et al. Induction of mesothelioma in p53+/− mouse by intraperitoneal application of multi-wall carbon nanotube. J Toxicol Sci. 2008;33(1):105–116. doi:10.2131/jts.33.105
15. Yang S-T, Wang X, Jia G, et al. Long-term accumulation and low toxicity of single-walled carbon nanotubes in intravenously exposed mice. Toxicol Lett. 2008;181(3):182–189. doi:10.1016/j.toxlet.2008.07.020
16. Al Rugaie O, Jabir MS, Mohammed MK, et al. Modification of SWCNTs with hybrid materials ZnO–Ag and ZnO–Au for enhancing bactericidal activity of phagocytic cells against Escherichia coli through NOX2 pathway. Sci Rep. 2022;12(1):17203. doi:10.1038/s41598-022-22193-1
17. Jwameer MR, Salman SA, Noori F, et al. Antiproliferative Activity of PEG-PEI-SWCNTs against AMJ13 breast cancer cells. J Nanomater. 2023;2023:1–8. doi:10.1155/2023/2855788
18. Ugarte D. Curling and closure of graphitic networks under electron-beam irradiation. Nature. 1992;359(6397):707–709. doi:10.1038/359707a0
19. Camisasca A, Giordani S. Carbon nano-onions in biomedical applications: promising theranostic agents. Inorg Chim Acta. 2017;468:67–76. doi:10.1016/j.ica.2017.06.009
20. Saifuddin N, Raziah A, Junizah A. Carbon nanotubes: a review on structure and their interaction with proteins. J Chem. 2013;2013:1–18. doi:10.1155/2013/676815
21. Pakhira B, Ghosh M, Allam A, Sarkar S. Carbon nano onions cross the blood brain barrier. RSC Adv. 2016;6(35):29779–29782. doi:10.1039/C5RA23534K
22. d’Amora M, Maffeis V, Brescia R, Barnes D, Scanlan E, Giordani S. Carbon nano-onions as non-cytotoxic carriers for cellular uptake of glycopeptides and proteins. Nanomaterials. 2019;9(8):1069. doi:10.3390/nano9081069
23. Miriyala N, Kirby DJ, Cumont A, et al. Synthesis of carbon onion and its application as a porous carrier for amorphous drug delivery. Crystals. 2020;10(4):281. doi:10.3390/cryst10040281
24. d’Amora M, Camisasca A, Boarino A, Arpicco S, Giordani S. Supramolecular functionalization of carbon nano-onions with hyaluronic acid-phospholipid conjugates for selective targeting of cancer cells. Colloids Surf B Biointerfaces. 2020;188:110779. doi:10.1016/j.colsurfb.2020.110779
25. Mamidi N, Delgadillo RMV, González-Ortiz A. Engineering of carbon nano-onion bioconjugates for biomedical applications. Mater Sci Eng C Mater Bio Appl. 2020;120:111698. doi:10.1016/j.msec.2020.111698
26. Mamidi N, Velasco Delgadillo RM, Gonzáles Ortiz A, Barrera EV. Carbon nano-onions reinforced multilayered thin film system for stimuli-responsive drug release. Pharmaceutics. 2020;12(12):1208. doi:10.3390/pharmaceutics12121208
27. Mamidi N, González-Ortiz A, Lopez Romo I, Barrera E. Development of functionalized carbon nano-onions reinforced zein protein hydrogel interfaces for controlled drug release. Pharmaceutics. 2019;11(12):621. doi:10.3390/pharmaceutics11120621
28. Frasconi M, Maffeis V, Bartelmess J, Echegoyen L, Giordani S. Highly surface functionalized carbon nano-onions for bright light bioimaging. Methods Appl Fluoresc. 2015;3(4):044005. doi:10.1088/2050-6120/3/4/044005
29. Lettieri S, Camisasca A, d’Amora M, et al. Far-red fluorescent carbon nano-onions as a biocompatible platform for cellular imaging. RSC Adv. 2017;7(72):45676–45681. doi:10.1039/C7RA09442F
30. Lettieri S, d’Amora M, Camisasca A, Diaspro A, Giordani S. Carbon nano-onions as fluorescent on/off modulated nanoprobes for diagnostics. Beilstein J Nanotechnol. 2017;8(1):1878–1888. doi:10.3762/bjnano.8.188
31. Ding L, Stilwell J, Zhang T, et al. Molecular characterization of the cytotoxic mechanism of multiwall carbon nanotubes and nano-onions on human skin fibroblast. Nano Lett. 2005;5(12):2448–2464. doi:10.1021/nl051748o
32. Xu Y, Wang SY, Yang J, et al. Multiwall carbon nano‐onions induce DNA damage and apoptosis in human umbilical vein endothelial cells. Environ Toxicol. 2011;28(8):442–450. doi:10.1002/tox.20736
33. Luszczyn J, Plonska‐Brzezinska ME, Palkar A, et al. Small noncytotoxic carbon nano‐onions: first covalent functionalization with biomolecules. Chemistry. 2010;16(16):4870–4880. doi:10.1002/chem.200903277
34. Bartelmess J, De Luca E, Signorelli A, et al. Boron dipyrromethene (BODIPY) functionalized carbon nano-onions for high resolution cellular imaging. Nanoscale. 2014;6(22):13761–13769. doi:10.1039/C4NR04533E
35. Marchesano V, Ambrosone A, Bartelmess J, et al. Impact of carbon nano-onions on Hydra vulgaris as a model organism for nanoecotoxicology. Nanomaterials. 2015;5(3):1331–1350. doi:10.3390/nano5031331
36. d’Amora M, Rodio M, Bartelmess J, et al. Biocompatibility and biodistribution of functionalized carbon nano-onions (f-CNOs) in a vertebrate model. Sci Rep. 2016;6(1):1–9. doi:10.1038/s41598-016-0001-8
37. Liu A, Wang C, Hehir M, Zhou T, Yang J. In vivo induction of CYP in mice by carbamazepine is independent on PXR. Pharmacol Rep. 2015;67(2):299–304. doi:10.1016/j.pharep.2014.10.002
38. Bland AR, Shrestha N, Rosengren RJ, Ashton JC. Does crizotinib auto-inhibit CYP3A in vivo? Pharmacology. 2020;105(11–12):715–718. doi:10.1159/000506996
39. Haider S, Liaquat L, Shahzad S, et al. A high dose of short term exogenous D-galactose administration in young male rats produces symptoms simulating the natural aging process. Life Sci. 2015;124:110–119. doi:10.1016/j.lfs.2015.01.016
40. Bray BJ, Goodin MG, Inder RE, Rosengren RJ. The effect of retinol on hepatic and renal drug-metabolising enzymes. Food Chem Toxicol. 2001;39(1):1–9. doi:10.1016/S0278-6915(00)00110-1
41. Deng X, Wu F, Liu Z, et al. The splenic toxicity of water soluble multi-walled carbon nanotubes in mice. Carbon. 2009;47(6):1421–1428. doi:10.1016/j.carbon.2008.12.032
42. Liu X, Zhang Y, Li J, et al. Cognitive deficits and decreased locomotor activity induced by single-walled carbon nanotubes and neuroprotective effects of ascorbic acid. Int J Nanomedicine. 2014;9:823. doi:10.2147/IJN.S56339
43. Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85. doi:10.1016/0003-2697(85)90442-7
44. Omura T, Sato R. A new cytochrome in liver microsomes. J Biol Chem. 1962;237(4):1375–1376. doi:10.1016/S0021-9258(18)60338-2
45. Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem. 1964;239(7):2370–2378. doi:10.1016/S0021-9258(20)82244-3
46. Nash T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem J. 1953;55(3):416. doi:10.1042/bj0550416
47. Kitada M, Igoshi N, Kamataki T, et al. Immunochemical similarity of P-450 HFLa, a form of cytochrome P-450 in human fetal livers, to a form of rat liver cytochrome P-450 inducible by macrolide antibiotics. Arch Biochem Biophys. 1988;264(1):61–66. doi:10.1016/0003-9861(88)90570-X
48. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi:10.1038/227680a0
49. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4354. doi:10.1073/pnas.76.9.4350
50. Kotegawa T, Laurijssens BE, Von Moltke LL, et al. In vitro, pharmacokinetic, and pharmacodynamic interactions of ketoconazole and midazolam in the rat. J Pharmacol Exp Ther. 2002;302(3):1228–1237. doi:10.1124/jpet.102.035972
51. Esterbauer H, Cheeseman KH. Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 1990;186:407–421.
52. Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44(1):276–287. doi:10.1016/0003-2697(71)90370-8
53. Seervi M, Lotankar S, Barbar S, Sathaye S. Assessment of cytochrome P450 inhibition and induction potential of lupeol and betulin in rat liver microsomes. Drug Metab Pers Ther. 2016;31(2):115–122. doi:10.1515/dmpt-2015-0043
54. Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270(1):414–423.
55. Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008;392(6):1093–1108. doi:10.1007/s00216-008-2291-6
56. Zuo G, Gu W, Fang H, Zhou R. Carbon nanotube wins the competitive binding over proline-rich motif ligand on SH3 domain. J Phys Chem C. 2011;115(25):12322–12328. doi:10.1021/jp2026303
57. Chauret N, Gauthier A, Nicoll-Griffith DA. Effect of common organic solvents on in vitrocytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab Dispos. 1998;26(1):1–4.
58. Wrighton SA, Ring BJ. Inhibition of human CYP3A catalyzed 1′-hydroxy midazolam formation by ketoconazole, nifedipine, erythromycin, cimetidine, and nizatidine. Pharm Res. 1994;11(6):921–924. doi:10.1023/A:1018906614320
59. Shvedova AA, Kisin E, Murray AR, et al. Inhalation vs. aspiration of single-walled carbon nanotubes in C57BL/6 mice: inflammation, fibrosis, oxidative stress, and mutagenesis. Am J Physiol Lung Cell Mol Physiol. 2008;295(4):L552–L565. doi:10.1152/ajplung.90287.2008
60. Patlolla AK, Berry A, Tchounwou PB. Study of hepatotoxicity and oxidative stress in male Swiss-Webster mice exposed to functionalized multi-walled carbon nanotubes. Mol Cell Biochem. 2011;358(1):189–199. doi:10.1007/s11010-011-0934-y
61. Shang S, Yang S-Y, Liu Z-M, Yang X. Oxidative damage in the kidney and brain of mice induced by different nano-materials. Front Biol. 2015;10(1):91–96. doi:10.1007/s11515-015-1345-3
62. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi:10.1155/2014/360438
63. Ighodaro O, Akinloye O. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alexandria J Med. 2018;54(4):287–293. doi:10.1016/j.ajme.2017.09.001
64. Tang AC, Hwang G-L, Tsai S-J, et al. Biosafety of non-surface modified carbon nanocapsules as a potential alternative to carbon nanotubes for drug delivery purposes. PLoS One. 2012;7(3):e32893. doi:10.1371/journal.pone.0032893
65. Shoji H, Miyakawa T. Differential effects of stress exposure via two types of restraint apparatuses on behavior and plasma corticosterone level in inbred male BALB/cAJcl mice. Neuropsychopharmacol Rep. 2020;40(1):73–84. doi:10.1002/npr2.12093
66. Liu Z, Davis C, Cai W, He L, Chen X, Dai H. Circulation and long-term fate of functionalized, biocompatible single-walled carbon nanotubes in mice probed by Raman spectroscopy. Proc Natl Acad Sci U S A. 2008;105(5):1410–1415. doi:10.1073/pnas.0707654105
67. Principi E, Girardello R, Bruno A, et al. Systemic distribution of single-walled carbon nanotubes in a novel model: alteration of biochemical parameters, metabolic functions, liver accumulation, and inflammation in vivo. Int J Nanomedicine. 2016;11:4299. doi:10.2147/IJN.S109950
68. Miyawaki J, Matsumura S, Yuge R, et al. Biodistribution and ultrastructural localization of single-walled carbon nanohorns determined in vivo with embedded Gd2O3 labels. ACS Nano. 2009;3(6):1399–1406. doi:10.1021/nn9004846
69. Li B, Zhang X-Y, Yang J-Z, et al. Influence of polyethylene glycol coating on biodistribution and toxicity of nanoscale graphene oxide in mice after intravenous injection. Int J Nanomedicine. 2014;9:4697. doi:10.2147/IJN.S66591
70. Choi SH, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. doi:10.1038/nbt1340
71. Elgrabli D, Dachraoui W, Ménard-Moyon C, et al. Carbon nanotube degradation in macrophages: live nanoscale monitoring and understanding of biological pathway. ACS Nano. 2015;9(10):10113–10124. doi:10.1021/acsnano.5b03708
72. Kagan VE, Kapralov AA, St. Croix CM, et al. Lung macrophages “digest” carbon nanotubes using a superoxide/peroxynitrite oxidative pathway. ACS Nano. 2014;8(6):5610–5621. doi:10.1021/nn406484b
73. Bussy C, Hadad C, Prato M, Bianco A, Kostarelos K. Intracellular degradation of chemically functionalized carbon nanotubes using a long-term primary microglial culture model. Nanoscale. 2016;8(1):590–601. doi:10.1039/C5NR06625E
74. Hou J, Wan B, Yang Y, Ren X-M, Guo L-H, Liu J-F. Biodegradation of single-walled carbon nanotubes in macrophages through respiratory burst modulation. Int J Mol Sci. 2016;17(3):409. doi:10.3390/ijms17030409
75. Valentine SP, Le Nedelec MJ, Menzies AR, Scandlyn MJ, Goodin MG, Rosengren RJ. Curcumin modulates drug metabolizing enzymes in the female Swiss Webster mouse. Life Sci. 2006;78(20):2391–2398. doi:10.1016/j.lfs.2005.09.017
76. Le Nedelec MJ, Rosengren RJ. Methylphenidate inhibits cytochrome P450 in the Swiss Webster mouse. Hum Exp Toxicol. 2002;21(5):273–280. doi:10.1191/0960327102ht245oa
77. Tang H-Q, Xu M, Rong Q, Jin R-W, Liu Q-J, Y-L L. The effect of ZnO nanoparticles on liver function in rats. Int J Nanomedicine. 2016;11:4275. doi:10.2147/IJN.S109031
78. Kulthong K, Maniratanachote R, Kobayashi Y, Fukami T, Yokoi T. Effects of silver nanoparticles on rat hepatic cytochrome P450 enzyme activity. Xenobiotica. 2012;42(9):854–862. doi:10.3109/00498254.2012.670312
79. Tenzer S, Docter D, Kuharev J, et al. Rapid formation of plasma protein Corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8(10):772–781. doi:10.1038/nnano.2013.181
80. Lunov O, Syrovets T, Loos C, et al. Differential uptake of functionalized polystyrene nanoparticles by human macrophages and a monocytic cell line. ACS Nano. 2011;5(3):1657–1669. doi:10.1021/nn2000756
81. Lesniak A, Salvati A, Santos-Martinez MJ, Radomski MW, Dawson KA, Åberg C. Nanoparticle adhesion to the cell membrane and its effect on nanoparticle uptake efficiency. J Am Chem Soc. 2013;135(4):1438–1444. doi:10.1021/ja309812z
82. Strojny B, Sawosz E, Grodzik M, et al. Nanostructures of diamond, graphene oxide and graphite inhibit CYP1A2, CYP2D6 and CYP3A4 enzymes and downregulate their genes in liver cells. Int J Nanomedicine. 2018;13:8561–8575. doi:10.2147/IJN.S188997
83. Schöttler S, Becker G, Winzen S, et al. Protein adsorption is required for stealth effect of poly (ethylene glycol)-and poly (phosphoester)-coated nanocarriers. Nat Nanotechnol. 2016;11(4):372–377. doi:10.1038/nnano.2015.330
84. Hadidi N, Shirazi SFH, Kobarfard F, Nafissi-Varchehd N, Aboofazeli R. Evaluation of the effect of PEGylated single-walled carbon nanotubes on viability and proliferation of Jurkat cells. Iran J Pharm Res. 2012;11(1):27–37.
85. Yang S, Guo W, Lin Y, et al. Biodistribution of pristine single-walled carbon nanotubes in vivo. J Phys Chem C. 2007;111(48):17761–17764. doi:10.1021/jp070712c
86. Zuo G, Huang Q, Wei G, Zhou R, Fang H. Plugging into proteins: poisoning protein function by a hydrophobic nanoparticle. ACS Nano. 2010;4(12):7508–7514. doi:10.1021/nn101762b
87. Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82(2):291–295. doi:10.1113/expphysiol.1997.sp004024
88. McCord JM. The evolution of free radicals and oxidative stress. Am J Med. 2000;108(8):652–659. doi:10.1016/S0002-9343(00)00412-5
89. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482(3):419–425. doi:10.1016/j.bbrc.2016.10.086
90. Sano N, Wang H, Chhowalla M, Alexandrou I, Amaratunga GA. Synthesis of carbon’onions’ in water. Nature. 2001;414(6863):506–507. doi:10.1038/35107141
91. Borgohain R, Yang J, Selegue JP, Kim DY. Controlled synthesis, efficient purification, and electrochemical characterization of arc-discharge carbon nano-onions. Carbon. 2014;66:272–284. doi:10.1016/j.carbon.2013.09.001
92. Huang W, Wang Y, Luo G, Wei F. 99.9% purity multi-walled carbon nanotubes by vacuum high-temperature annealing. Carbon. 2003;41(13):2585–2590. doi:10.1016/S0008-6223(03)00330-0
93. Dillon AC, Gennett T, Jones KM, Alleman JL, Parilla PA, Heben MJ. A simple and complete purification of single‐walled carbon nanotube materials. Adv Mater. 1999;11(16):1354–1358. doi:10.1002/(SICI)1521-4095(199911)11:16<1354::AID-ADMA1354>3.0.CO;2-N
94. Shvedova A, Castranova V, Kisin E, et al. Exposure to carbon nanotube material: assessment of nanotube cytotoxicity using human keratinocyte cells. J Toxicol Environ Health A. 2003;66(20):1909–1926. doi:10.1080/713853956
95. Ge C, Li Y, Yin -J-J, et al. The contributions of metal impurities and tube structure to the toxicity of carbon nanotube materials. NPG Asia Mater. 2012;4(12):e32. doi:10.1038/am.2012.60
96. Tan YZ. Toxicological Assessment of Carbon Nano-Onions in BALB/c Mice [Thesis, Master of Science]. University of Otago; 2022.



